
Abstract
Water deficit is a major constraint for crops of economic importance in almost all agricultural regions. However, plants have an active defense system to adapt to these adverse conditions, acting in the reprogramming of gene expression responsible for encoding microRNAs (miRNAs). These miRNAs promote the regulation to the target gene expression by the post-transcriptional (PTGS) and transcriptional gene silencing (TGS), modulating several pathways including defense response to water deficit. The broader knowledge of the miRNA expression profile and its regulatory networks in response to water deficit can provide evidence for the development of new biotechnological tools for genetic improvement of several important crops. In this study, we used Setaria viridis accession A10.1 as a C4 model plant to widely investigate the miRNA expression profile in early responses to different levels of water deficit. Ecophysiological studies in Setaria viridis under water deficit and after rewatering demonstrated a drought tolerant accession, capable of a rapid recovery from the stress. Deep small RNA sequencing and degradome studies were performed in plants submitted to drought to identify differentially expressed miRNA genes and their predicted targets, using in silico analysis. Our findings showed that several miRNAs were differentially modulated in response to distinctive levels of water deficit and after rewatering. The predicted mRNA targets mainly corresponded to genes related to cell wall remodeling, antioxidant system and drought-related transcription factors, indicating that these genes are rapidly regulated in early responses to drought stress. The implications of these modulations are extensively discussed, and higher-effect miRNAs are suggested as major players for potential use in genetic engineering to improve drought tolerance in economically important crops, such as sugarcane, maize, and sorghum.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Climate change has become increasingly global and accentuated, imposing strong evolutionary challenges for plants (Burney and Ramanathan 2014; Lawrence and Vandecar 2015; Rosenzweig et al. 2014). Water deficit is one of the most severe abiotic stresses that impairs several economically important crops, negatively impacting growth and productivity, or preventing their cultivation in regions with prolonged drought period (Farooq et al. 2009). However, plants have their own defense systems to adapt to adverse weather conditions (Liu et al. 2016). A particular mechanism is the reprogramming of gene expression encoding microRNAs (miRNAs) (Ferdous et al. 2015a; Guerra et al. 2015). Recent studies have provided evidence that miRNAs are indeed drought-responsive (Ferdous et al. 2015a; Hackenberg et al. 2015) and play critical roles in modulation of the several mechanisms of adaptation and tolerance to water deficit in plants (Liu et al. 2016).
The miRNAs are described as non-coding small RNAs with typically 21–24 nucleotides (nt) and are encoded by genes located in both the intergenic and intragenic regions. The action of a miRNA is based on perfect or partial complementary base pairing between it and one or more target messenger RNA (mRNA) molecules, enabling the the modulation of gene expression by mRNA site-specific cleavage, deacetylation of the poly(A) tail or steric inhibition of the gene translation process (Post-Transcriptional Gene Silencing pathway; PTGS) (Pacak et al. 2015; Borges and Martienssen 2015; Ferdous et al. 2015a; Guerra et al. 2015; Hackenberg et al. 2015; Jeong et al. 2011). In addition, they also act in transcriptional gene silencing (TGS; RNA-directed target DNA methylation) and targeted methylation of genomic regions (introns, exons or promoter sequences) to affect gene expression (Matzke and Mosher 2014; Matzke et al. 2015). miRNA deep sequencing (small RNAseq analysis) (Lu et al. 2007) and analysis of RNA ends resulting from miRNA cleavage using a high-throughput deep sequencing method (Addo-Quaye et al. 2008), stem-loop RT-qPCR (Chen et al. 2005) followed by real time RT-qPCR has proven to be an important tool that has been used to identify and quantify the expression of a given miRNA and its target mRNA in several biological systems (Basso et al. 2019).
The knowledge of the miRNA expression profile and its regulatory networks in plants under water deficit may provide important information to deepen our understanding of molecular mechanisms involved in defense response. Several miRNAs and their targets associated with abiotic stress responses have been identified using overexpression, silencing, and mutation techniques in plant species including sugarcane (Wang et al. 2022), maize (Fatma and Aydinoglu 2020), rice (Yue et al. 2017) and millets (Chakraborty et al. 2020). A profiling of the salt stress responsive miRNAs in Setaria viridis identified an abundance of alterations of miRNAs, where miR169, miR395, miR396, miR397, miR398 and miR408 contributed for elucidation of the regulatory network in salt stress response (Pegler et al. 2020). In Setaria italica (foxtail millet), Wang et al. (2016) identified several miRNAs under drought conditions. A more recent study, conducted by Chakraborty et al. (2020), used five millet species to identify miRNAs conserved within them, in addition to novel micro RNAs potentially involved in drought response. The overexpression in Arabidopsis thaliana of Sit-miR394 indicated an increase in tolerance to drought stress, suggesting that Sit-miR394 is involved in the response to drought (Geng et al. 2021). Furthermore, these findings can contribute to the development of biotechnology tools focused on plant genetic improvement aimed at drought tolerance (Hajyzadeh et al. 2015; Kim et al. 2009; Ni et al. 2012; Song et al. 2013; Zhou et al. 2013).
Here, we used Setaria viridis as a model plant to extensively investigate the miRNA expression profile in early responses to drought and after rehydration. S. viridis is a diploid, C4 monocot grass that belongs to the Poaceae family and is commonly used as a model plant for grasses (Brutnell et al. 2015, 2010; Ermawar et al. 2015; Li and Brutnell 2011). In fact, this model plant has been widely used for functional genomics and proof of concepts in bioenergy feedstocks and panicoid food crops (Brutnell et al. 2015, 2010; Huang and Brutnell 2016; Li and Brutnell 2011; Ribeiro et al. 2017).
In this study, we aim to evaluate the expression profile of drought responsive miRNA genes, and to identify their target mRNAs in plants subjected to different water levels in the soil. Small RNA and degradome sequencing from A10.1 accession using Illumina platforms were performed, resulting in small RNAseq and degradome sequencing libraries. The expression profile of the conserved miRNAs retrieved from miRBase database (Kozomara and Jones 2014) and novel miRNA identified by in silico prediction from this study were evaluated. Our findings showed that several miRNA genes were differentially modulated in early responses to decreasing levels of available water. The implications of these modulations are extensively discussed, and higher-effect miRNAs are highlighted as major players for potential use in genetic engineering to improve drought tolerance in economically important crops.
Materials and methods
Plant and growth conditions
Seeds of wild-type S. viridis accessions A10.1 were treated with concentrated sulfuric acid to promote dormancy break. After blotted on sterile filter paper, seeds were placed on sterile 1/2 Murashige and Skoog (MS) agar media (Murashige and Skoog 1962) and incubated for seven days in a growth chamber (Conviron®) under 16 h photoperiod with 150 µmol m−2 s−1 light intensity and 25 °C. Seedlings were transplanted and acclimated in pots containing 250 g of soil, substrate (Plantmax®) and vermiculite in a mixture of 3:1:0.5 (v/v/v). Plants were maintained in the growth chamber (Fitotron®) under 16 h photoperiod with 500 µmol m−2 s−1, 26 °C and 65% relative humidity.
Setting of water deficit levels and time of plant sampling
The soil hydric potential (Ѱw) was previously determined by a water retention curve using a psychrometer WP4C Dewpoint PotentiaMeter (Decagon WP4, Pullman, WA, USA). Four levels of available water in the soil were used: 0% (Ѱw -1.50 MPa; permanent wilting point or PWP), 25% (Ѱw − 1.125 MPa; severe deficit), 50% (Ѱw − 0.75 MPa; moderate deficit) and 100% (Ѱw − 0.03 MPa; field capacity or FC). Water deficit treatments were applied in adult plants in the onset of flowering, which were maintained in the growth chamber (Fitotron®) in the same conditions described above. Moreover, an additional plant set maintained at − 1.4 MPa (right before PWP) for 24 h was rewatered to FC (rewatering or rehydration; RW) and evaluated after 24 h. All samples from each treatment (0, 25, 50 and 100%) were collected 24 h after the onset of the stress, while the rehydrated plant tissues were collected 24 h after rewatering. The trials were carried out in a randomized block design with three biological replicates for both drought treatments and each biological replicate was composed of six plants.
Ecophysiology analysis
Gas exchange measurements were carried out using a conventional infrared gas analyzer system with a 6 × 6 cm2 clamp-on leaf cuvette (LICOR, LI-6400). Photosynthetic photon flux density (PPFD) was fixed at 1500 μmol m−2 s−1, using a red-blue LED light source built into the leaf cuvette. The vapor pressure deficit in the cuvette was maintained below 2.5 kPa to prevent stomatal closure due to the low air humidity effect. Carbon dioxide was supplied from a high-pressure liquefied CO2 cylinder, with mean CO2 concentration of 400 μmol mol−1. All measurements were performed in the leaves + 2 (younger and fully expanded leaf) with three biological replicates and six plants in each replicate. Net photosynthetic rate (A), stomatal conductance (gs), leaf transpiration rate (E), intercellular CO2 and water-use efficiency (WUE) were measured. Leaf water potential (ΨL) and osmotic potential (Ψs) were measured from three plants from each experimental plot (six drought treatments, three biological replicates each and three plant for each replicate), using a thermocouple psychrometry with a 14 channel Wescor/Campbell Water Potential System [C-52 sample chambers, plus the CR7 Measurement and 700X Control System]. Leaf discs of 0.5 cm2 from leaves + 2 were placed in the sampling chamber and analyses were performed according to standard procedure.
RNA isolation, cDNA synthesis and RT-qPCR
The same leaves + 2 used for physiological measurements were transferred to liquid nitrogen and stored at − 80 °C until analyses. Total RNA was isolated using TRIzol reagent according to the manufacturer’s instructions. RNA samples were treated with RNase-free RQ1 DNase I (Promega) according to the manufacturer’s instructions. Then, 2 μg of RNA were used as template for cDNA synthesis using Oligo-(dT)20 primer and SuperScript III RT, according to the manufacturer’s instructions. Reactions were performed using 200 ng of cDNA, 0.2 µM of each gene-specific primer and Platinum™ SYBR™ Green qPCR SuperMix-UDG with ROX, in a final volume of 10 μL. All samples were carried out in technical triplicate. Primers efficiencies and target-specific amplification were confirmed by a single and distinct peak in melting curve analysis. Thermocycling conditions were performed according to Martins et al. (2016). The relative expression levels of the genes were analyzed by the 2−∆∆Ct method (Schmittgen and Livak 2008), using translation factor SUI1 (SvSUI; Sevir.2G348300) as an endogenous reference gene. Previous molecular characterization of the plant set submitted to different levels of water deficit was performed based on the expression profile of delta-1-pyrroline-5-carboxylate synthetase (SvP5CS; Sevir.5G386300) and galactinol synthase 2 (SvGolS2; Sevir.2G450900) genes. All assays were performed using three biological replicates with three technical replicates each and a non-template control. All primers used are listed in Table S1.
Small RNA isolation and NGS
Small RNAs were isolated from 16 samples of control/treated plants using mirVana miRNA Isolation Kit, according to manufacturer's instructions. Then, 10 µg from each sample were homogenized and dried in GenTegra RNA tubes, according to manufacturer's instructions. Libraries were prepared with small RNAs of 15–36 nucleotides (nt) in length retrieved from 15% polyacrylamide gel and using TruSeq small RNA SBS kit v.3, according to manufacturer's instructions. NGS was performed using the Illumina HiSeq 2500 platform, generating 15 libraries, which were demultiplexed using CASAVA v.1.8.4 pipeline, allowing one mismatch in the index selection.
Stem-loop RT-qPCR and RT-qPCR of predicted miRNA targets
One differentially expressed miRNA gene was chosen and validated. RNA samples treated with DNase I were reverse transcribed (RT) by the pulsed method using SuperScript III RT and stem-loop adaptors/primers (1 µM each), and Oligo-(dT)20 primers (100 µM) were used for internal controls in each single RT reaction. The samples were incubated at 16 °C for 30 min followed by pulsed RT of 60 cycles at 30 °C for 30 s, 42 °C for 30 s and 50 °C for 1 s. Then the reaction was stopped by incubating the samples at 85 °C for 5 min as described by Gasic et al. (2007). Thermocycling conditions were carried out according to Ferdous et al. (2015b). The relative expression levels of these miRNAs were analyzed by the 2−∆∆Ct method (Schmittgen and Livak 2008) using GAPDH as an endogenous reference gene. The miRNA specific stem-loop RT primers were designed following the method described by Chen et al. (2005) and Gasic et al. (2007). All primers used are listed in Table S1.
NGS data mining
Information present in the supplemental methods file.
Degradome sequencing of plants submitted to severe stress
Information present in the supplemental methods file.
Degradome data mining
Information present in the supplemental methods file.
Results
Assessment of physiological and molecular responses to different levels of soil water content in S. viridis
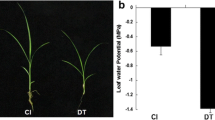
To assess the responses of S. viridis to drought stress, we performed molecular and ecophysiology studies in plants under different levels of water deficit and subsequent rewatering (RW). Four levels of soil water were applied (Fig. 1A): 100%, 50%, 25%, and 0%. An additional set of plants was also evaluated after rewatering, which was carried out in plants maintained at− 1.4 MPa for 24 h. Previously, the soil hydric potential (Ѱw) was determined by a water retention curve (Fig. 1B).

Phenotyping of Setaria viridis under different levels of water deficit. a Setaria viridis (accession A10.1) maintained under available water levels of 100%, 50%, 25% and 0%. Rewatering consists of plants maintained at − 1.4 MPa for 24 h and then rewatered to field capacity, which were evaluated 24 h after rehydration. Photographs were taken in two stages: 24 h after the onset of plant stress and 24 h after rewatering. b Matrix water retention curve to determine the relationship between water levels and water soil potential (MPa). c Expression profile of the drought molecular marker genes SvP5CS and SvGolS2. Letters above the bars indicate statistically significant difference between different conditions (P-value < 0.05, Tukey's HSD test)
The molecular characterization of plants submitted to different levels of soil water content was performed based on the expression profile of delta-1-pyrroline-5-carboxylate synthetase (SvP5CS; Sevir.5G386300) and galactinol synthase 2 (SvGolS2; Sevir.2G450900) genes. As observed in Fig. 1C, both SvP5CS and SvGolS2 were up-regulated in plants under water deficit, especially in severe stress conditions. The rewatering of the plants drastically decreased the expression levels of these genes, suggesting a rapid osmotic adjustment after plant recovery from drought.
The ecophysiological analyses of plants submitted to drought stress and after recovery is demonstrated in Fig. 2. Gas exchange measurements demonstrated the robustness of the drought stress assays, as the CO2 assimilation (A), stomatal conductance (gs) and the transpiration rate (E) decreased proportionally to the water level applied. Water use efficiency (WUE), determined by A/E relation, increased under moderate and severe stress, a response that is typical of photosynthetically efficient C4 plants. The leaf water potential and the osmotic potential also decreased proportionally to decreased water levels. After rewatering, the plants were able to restore the physiological levels of A, E, gs leaf water and osmotic potential, a response indicative of drought-tolerant plants (Fig. 2).

Changes in physiological parameters of Setaria viridis under different levels of water deficit. Water and osmotic potentials, CO2 assimilation (A), transpiration rate (E) and stomatal conductance (gs), water use efficiency (WUE), were measured as described in materials and methods. The measurements were taken 24 h after the onset of drought stress or rewatering. Available water levels used were 100%, 50%, 25% and 0%. Rewatering (red squares) consisted of plants maintained at − 1.4 MPa (0% soil water level, right before PWP) for 24 h and then rehydrated to field capacity
Overview of sRNAs in S. viridis under drought stress
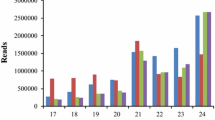
To identify differentially expressed miRNA genes in S. viridis under different levels of drought stress, small RNA deep sequencing was performed. The quality of the pooled RNA from each library and the quality of the downstream analysis were assessed as described in the materials and methods of this manuscript. An overview of the raw data quality and amount of total and unique reads from trimmed and filtered (18 to 26 nucleotides length) libraries is shown in Table S2. It was possible observe a similar read distribution of lengths in all libraries (Fig. 3A and 3B), with the 24 nt reads being the most abundant in total (~ 350,000 RPM) and unique reads (~ 150,000 RPM), followed by the 22 nt in total (~ 250,000 RPM) and 23 nt in unique reads (~ 20,000 RPM). The total and unique reads, as the number of reads successfully mapped to S. viridis genome in each library are presented in Supplementary File 1. For subsequent analysis, only matching reads to Setaria were used.

Amount of total a and unique b reads (18 to 26 nucleotides length) per treatment from normalized data (reads per million; RPM). c Venn diagrams showing overlapping of differentially expressed conserved and novel miRNAs genes in plants submitted to different water deficit levels and after rewatering (RW). The miRNA genes up- or down-regulated by the water deficit or after rewatering compared to control plants were determined when fold-change of magnitude < − 0.8 or > 0.8 and P-value < 0.05 was observed
Identification and expression profile analysis of conserved and novel miRNAs in S. viridis submitted to different levels of drought stress
To identify conserved and novel miRNAs in S. viridis, filtered reads from RNA sequencing were mapped against conserved mature miRNAs retrieved from miRBase 22.1 database (Kozomara et al. 2019) and SIFGD Database (You et al. 2015). Reads unmapped in these miRNAs were used to predict novel miRNA using miRCat algorithm (Stocks et al. 2012). A total of 302 conserved and 753 novel miRNAs, comprising 65 different families, were identified in S. viridis (Supplementary Files 2 and 3).
The number of reads mapped without mismatches in conserved and predicted mature miRNAs was used for the analysis of differentially expressed genes (DEGs), based on the number of occurrences of its respective reads/sequence in each library. As shown in Fig. 3C and in Tables 1 and 2, a total of 31 conserved miRNAs were differentially expressed under moderate stress compared to control conditions, while 18 and 26 conserved miRNAs were differentially expressed under severe stress and the PWP, respectively. In addition, a total of 34, 19 and 42 novel miRNAs were differentially expressed under moderate, severe and the PWP conditions, respectively, compared to control plants. After rewatering, 17 conserved and 14 novel miRNAs genes were differentially expressed compared to control plants. A total of 53 miRNA families are identified in moderate stress, while 34 and 62 families in severe and PWP, respectively, and 29 in re-watering (Table S3). Venn diagrams showing the number of overlapped differentially expressed miRNAs genes in the different conditions tested are shown in Fig. 3C.
The expression profile of conserved and novel DEGs of miRNAs is presented in Tables 1 and 2 and Supplementary Files 4 and 5. The expression profile of the miRNA genes appears to be highly dynamic, changing according to the level of water deficit applied. Some miRNA genes are exclusively up- or down-regulated only in drought stress conditions but are not significantly expressed after plant rehydration. Among the conserved miRNA genes exclusively up-regulated in drought stress (moderate, severe and at the PWP) were aly-miR165b-3p, gma-miR171t, gma-miR6300, mes-miR535a, stu-miR171b-3p, stu-miR172c-3p, zma-miR164f-5p and zma-miR172d-3p, while the exclusively down-regulated miRNA genes in all stress conditions were ppt-miR894, tcc-miR396d and zma-miR396f-5p (Table 1). The novel miRNA genes exclusively up-regulated during the stress conditions but not after rewatering comprised SvmiRp175, SvmiRp351, SvmiRp571, SvmiRp605, SvmiRp620 and SvmiRp629, while the exclusively down-regulated novel miRNA gene in these conditions was SvmiRp108 (Table 2). After rewatering, nine exclusive conserved miRNA genes were up-regulated, with nov-sit-miR96, osa-miR408-5p and zma-miR528-3p being extremely up-regulated (log2-fold > 2.0), in addition to six conserved miRNA genes only down-regulated during rehydration, including nov-sit-miR143, Sit-miR5568.20, Sit-miR5568.74, vvi-miR171f, zma-miR167g-3p and zma-miR319d-3p (Table 1). Five novel miRNA genes were exclusively up-regulated after rewatering, SvmiRp330, SvmiRp422, SvmiRp734, SvmiRp942 and SvmiRp969, and two novel miRNA genes were exclusively down-regulated after rehydration, SvmiRp612 and SvmiRp952 (Table 2).
The miRNA genes differentially expressed only during moderate stress (50% water level) or both severe stresses (25% water level and PWP) could also be identified. The conserved miRNA genes only up-regulated during moderate stress were lus-miR166b, nov-sit-miR49, osa-miR444c.1, ppe-miR171h, stu-miR399i-3p, stu-miR408b-3p, tcc-miR530a, zma-miR171j-3p and zma-miR399j-3p, while the conserved up-regulated miRNA genes only in severe stress conditions were zma-miR159a-5p and zma-miR167g-3p (Table 1). In contrast, the conserved miRNA genes only down-regulated during moderate stress were ata-miR393-3p, bdi-miR159a-3p, osa-miR162b and zma-miR159k-3p, while the exclusively down-regulated conserved miRNA genes in severe stress were nov-sit-miR111, osa-miR408-5p and zma-miR528b-3p.
It is important to highlight that two conserved miRNA genes were down-regulated in severe conditions and up-regulated during rewatering (osa-miR408-5p and zma-miR528b-3p), while zma-miR167g-3p demonstrated the reversed trend, as it was up-regulated in severe stress and down-regulated during rewatering (Table 1). These results suggest that these miRNAs could be important for the regulation of genes on stress recovery.
Regarding novel miRNA genes' differential expression exclusively in moderate or severe stresses we can highlight SvmiRp80, 453, 466, 471, 730, 751 and 826 being up-regulated only during moderate stress, while SvmiRp153 and 405 were up-regulated only in severe stress. Moreover, we also found the novel miRNA gene SvmiRp10 as being down-regulated only in moderate stress while SvmiRp86, 137 and 402 genes were down-regulated exclusively in severe stress conditions. An illustrative representation of the results described above is demonstrated in Fig. 4 and a summary of the predicted gene targets of these differentially expressed miRNAs in each condition can be found in Supplementary File 6.

Overview of differentially expressed miRNA genes and expression levels of the miRNA/target gene pairs by RT-qPCR. a Illustrative representation of differentially expressed conserved or novel miRNA genes in Setaria viridis submitted to different levels of water deficit (moderate or severe). I, II and III represent well-hydrated plants, moderate and severe drought stress, respectively. The red and green colors indicate the genes down- or up-regulated in each condition, respectively. Differentially expressed genes (DEGs) in severe stress correspond to DEGs observed in plants both under 25% and 0% of soil water content. b Expression levels of miRNA genes and their predicted targets. The heatmap represents the fold-change in expression levels, calculated according to the 2−∆∆Ct method (Schmittgen and Livak 2008). Each color in the heatmap indicate a different miRNA/target gene module
GO analysis and target predictions
We predicted a total of 5846 and 794 novel and conserved miRNA target pairs, respectively, and a full list of these pairs is presented in Supplementary Files 7 and 8. GO analysis of differentially expressed conserved and novel miRNA genes suggested that the enriched cellular components included membrane- and nuclear-related genes, whereas in the molecular function and biological process categories binding/DNA binding and metabolic and biosynthetic processes were enriched both in stress conditions and rewatering (Fig. 5). It is interesting to note that many categories are exclusively enriched during rewatering, indicating the plasticity of S. viridis to recover from drought stress.

Gene Ontology (GO) analysis from predicted target genes of all (conserved and novel) differentially expressed miRNAs in response to water deficit or rewatering. The enriched cellular components included membrane- and nuclear-related genes, whereas in the molecular function and cellular component
Using in silico analysis, we were able to predict target mRNAs of the DEG miRNAs (Supplementary Files 6, 7 and 8). Predictive analyzes of the novel miRNA targets are complicated to perform, since it was found that a single novel miRNA can regulate many different genes (Supplementary File 7). However, it is noteworthy that a significant number of predicted targets for novel DEGs of miRNAs are cell wall-related genes (Supplementary File 6). A detailed analysis of miRNA DEGs associated with their predicted targets reveals a complex regulation of drought responses observed in S. viridis, which is highly dependent on the level of stress applied (Fig. 4A; Tables 1 and 2; Supplementary File 6 and Supplementary Fig. S1).
In moderate drought stress, osa-miR444c.1, a miRNA possibly regulating two MADS-box transcription factor genes (MADS27; Sevir.7G133400 and MADS57; Sevir.1G314300) was up-regulated, while bdi-miR159a-3p, osa-miR162b and zma-miR159k-3p, possibly targeting different MYB family transcription factors, were down-regulated. Regarding novel miRNAs, it was observed that SvmiRp826, predicted to regulate a MADS25 gene (Sevir.5G034800), was also up-regulated exclusively during moderate drought stress, indicating that MADS-box transcription factor genes could have an important role in these conditions. During moderate stress, two miRNA genes, bdi-miR159a-3p and zma-miR159k-3p, were down-regulated (Fig. 4A; Table 1; Supplementary File 6 and Fig. S1). The predicted targets of these miRNAs belong to a wide range of different MYB family transcription factors (Sevir.5G360500, Sevir.1G076400, Sevir.9G180500 and Sevir.3G193000).
Some conserved miRNA genes demonstrated differential responses during severe stress and after rewatering, suggesting an important role of these miRNA genes in S. viridis recovery from drought. Some examples included in this classification are osa-miR408-5p, zma-miR528b-3p, zma-miR167g-3p and nov-sit-miR111 (Fig. 4A; Table 1 and Supplementary File 6). The miRNA gene osa-miR408-5p is predicted to target a stress responsive protein gene (Sevir.2G448800), two chalcone synthase genes (Sevir.7G023500 and Sevir.7G023600), a N-acetyltransferase 9 gene (Sevir.9G057500) and a putative lipid transfer protein gene (Sevir.9G057600). The miRNA zma-miR528b-3p is predicted to regulate Sevir.5G176900 and the zma-miR167g-3p gene is predicted to putatively target four uncharacterized genes: Sevir.1G201700, Sevir.3G196500, Sevir.3G231500 and Sevir.7G200100. The nov-sit-miR111 is predicted to target an ethylene-insensitive protein gene (Sevir.9G126600), and a novel miRNA gene, SvmiRp86, down-regulated exclusively in severe stress, is also predicted to target an ethylene-insensitive protein gene (Sevir.2G264700).
Identification of miRNA target genes by degradome sequencing
The degradome sequencing was applied to identify novel and conserved miRNA targets in S. viridis submitted to severe water stress. A total of 15.232.695 raw reads were obtained and mapped against the S. viridis transcriptome (Bennetzen et al. 2012). After mapping, 10.759.326 (70.63%) matched tags and 4.058.119 unique mappable reads were obtained. The unique mappable reads were cleaned and an output of 3.55.409 clean tags obtained. The clean tags mapped were used as input in CleaveLand v3.0.1 pipeline to predict cleaved sites in S. viridis miRNA (Addo-Quaye et al. 2009).
In S. viridis degradome results, we found 482 cleavage targets associated with 130 miRNAs and 307 target genes classified in five categories (0–4). The categories are separated according to the relative abundance of tags in mRNA target sites with a P‐value < 0.05 (Supplementary File 9). Categories 0 and 1 are weighted greater due to a higher number of reads at the target position (Addo-Quaye et al. 2009). Among the identified targets, 18, 122, 151, 1 and 189 targets were detected as belonged to categories 0, 1, 2, 3 and 4, respectively. The results indicated that predicted targets interfere in the regulation of a wide range of biological process, including transcription factors, amino acid transport, posttranslational modification, protein turnover, chaperones, carbohydrate transport, translation, ribosomal structure, biogenesis, intracellular trafficking, secretion, energy production/conversion, vesicular transport, signal transduction mechanism, lipid transport, coenzyme transport, cell wall/membrane/envelope biogenesis, replication, recombination, repair, energy production and defense mechanisms.
As expected, the results showed that a unique miRNA is capable of cleaving several target genes, but also one target might be cleaved by different miRNAs. Some of these miRNA target genes and transcription factors were associated with drought and dehydration stress responses. These transcripts and genes included NAC transcription factors (far-miR164a/far-miR164b/Sevir.3G402300), genes associated with amino acid metabolism (ggo-miR-548a/hsa-miR-548ad-5p/hsa-miR-548ae-5p/hsa-miR-548ay-5p/hsa-miR-548d-5p/ppy-miR-548a/Sevir.3G024800, nta-miR6145d/nta-miR6145e/Sevir.1G359000 and tae-miR9777/Sevir.9G415200) and signal transduction SAPK-related genes (bmo-miR-2742/Sevir.5G029800, cel-miR-261/Sevir.2G037000 and tca-miR-3859-5p/Sevir.3G235900).
Two novel miRNAs candidate targets in S. viridis under severe drought stress corresponded to E3 ubiquitin ligase SUD1 gene (SvmiRp179/Sevir.4G213600) and ADP-ribosylation factor GTPase-activating protein AGD12-like gene (SvmiRp537/Sevir.2G335000). Consensus results of the degradome and small RNAseq (miRNA DEGs) in severe stress reveal only one miRNA (osa-miR408-5p) common to both analyses (Supplementary Files 8 and 9). This miRNA, which was down-regulated in severe stress and up-regulated during rewatering, putatively regulates a stress responsive protein gene (Sevir.2G448800), reinforcing that miRNA regulation of this gene could be important to S. viridis recovery after severe drought. Hence, we validated osa-miR408-5p expression levels observed from the sRNA sequencing data and the potential correlation between this miRNA and its predicted target Sevir.2G448800 (Fig. 4B and Supplementary Fig. 1).
Validation of miRNAs and their predicted targets gene expression
The qPCR results demonstrated that the expression levels of osa-miR408-5p, bdi-miR159a-3p, zma-miR396f-5p and SvmiRp86 genes corresponded to the expression profile observed for the different water deficit conditions (Fig. 4B; Fig. S2; Tables 1 and 2). In addition, the expression levels of their predicted targets were negatively correlated with the respective miRNA gene expression pattern, further corroborating the potential involvement of these modules in responses of S. viridis to drought.
The osa-miR408-5p gene was down-regulated in severe stress (Table 1; Fig. 4; Supplementary Fig. S2), while its predicted target, a stress-responsive protein gene (SSR), was up-regulated compared to control plants (Fig. 4B; Fig. S2). The bdi-miR159a-3p gene was down-regulated in drought stress compared to control conditions (Table 1; Fig. 4), while the expression of its predicted targets, Sevir.5G360500 and Sevir.1G076400, both corresponding to the MYB transcription factor gene family, were negatively correlated (Fig. 4B; Fig. S2). Two growth regulator factor genes, Sevir.8G160800 and Sevir.9G140200, were down-regulated while their target miRNA, zma-miR396f-5p was up-regulated in drought conditions (Table 1; Fig. 4B; Fig. S2). Furthermore, we have chosen to validate a novel S. viridis miRNA gene, SvmiRp86, which was down-regulated during severe stress when compared to hydrated plants and demonstrated a variety of predicted targets (Supplementary File 6). As most of these targets correspond to cell wall-related genes, we were able to verify that the expression of four genes, Sevir.4G159200 (1,4-alpha glucan), Sevir.9G155700 (expansin), Sevir.2G281200 (beta-glucosidase) and Sevir.7G167900 (UDP-glucuronosyl/UDP-glucosyltransferase), all related to cell wall-modifying genes, was negatively correlated with SvmiRp86 expression (Fig. 4; Fig. S2), suggesting that these modules participate in cell wall responses to drought in S. viridis.
Discussion
MicroRNAs modulate the expression of target mRNAs and regulate several plant processes, including biotic and abiotic stress responses. Understand the fine-tuning of miRNA genes is an important strategy to increase the crop's tolerance to abiotic or biotic stresses (Basso et al. 2019). Setaria viridis is emerging as a model plant for C4 grasses, and recent studies have demonstrated that phenotypic alterations in S. viridis using biotechnological approaches could be translated to sugarcane (Saccharum spp.), an important bioenergy crop (de Souza et al. 2018; 2019). Here, we aimed to study the fine-tuning of miRNA genes during early responses of S. viridis submitted to different levels of drought stress (moderate and severe drought stress and rewatering).
Physiological and molecular responses of S. viridis to drought stress
The robustness of the drought stress applied could be verified by ecophysiological studies demonstrating that the photosynthesis (A), stomatal conductance (gs) and the transpiration rate (E) decreased proportionally to the soil water content (Fig. 2). After rewatering of plants submitted for 24 h to − 1.40 MPa, the plants were able to restore the physiological levels of A, E, gs leaf water and osmotic potential to levels similar to control plants (Fig. 2). Moreover, the molecular markers for drought stress (SvP5CS and SvGolS genes responsible for osmolyte accumulation) were up-regulated during severe stress and down-regulated after rewatering, indicating a rapid osmotic adjustment in S. viridis. Overall, plants under severe water deficit shows 40–60% of the highest photosynthetic rate 24 h after rewatering, and the recovery occur in the subsequent days, regardless of the fact that full photosynthesis rates are not always reestablished (Frosi et al. 2017). Altogether, our results reinforced a study performed by Saha et al. (2016), which demonstrated that S. viridis (accession A10.1) is a drought tolerant plant, and therefore is an interesting model for prospecting drought tolerant-related genes.
Differential drought-responsive miRNA
The distinctive levels of soil water content led to a differential miRNA gene expression profile in leaves of S. viridis, which was highly dependent on the severity of the drought stress applied. Our experimental conditions allowed us to detect early responses to drought and rewatering, as the analyses were performed only 24 h after the onset of each stress or rehydration.
Moderate drought-responsive miRNA profile
During moderate drought stress, the differentially expressed genes of miRNAs (DEG miRNAs) included the up-regulated genes osa-miR444c.1, stu-miR408b-3p and tcc-miR530a, and the down-regulated bdi-miR159a-3p and zma-miR159k-3p genes. The predicted targets of osa-miR444c.1 comprise two genes related to MADS-box family genes, OsMADS27 and OsMADS57, which are members of AGL17-like clade of transcription factors known to be involved in stress responses (Arora et al. 2007). The up-regulation of osa- miR444c.1 indicates that OsMADS27 and OsMADS57 could be silenced during moderate drought. In rice, OsMADS27 was not responsive to moderate osmotic stress, while OsMADS57 was down-regulated in these conditions (Puig et al. 2013). In Arabidopsis, miR156 overexpression was shown to silence a MADS-box protein (AGL79), to modulate leaf morphology and lateral root development (Gao et al. 2018). Comp6823, which encodes for a MADS-box gene in C. colocynthis, is up-regulated markedly only at final stages of drought in this species (Wang et al. 2014). These results suggest that some MADS-box genes are turned off during early stages of drought. In contrast, bdi-miR159a-3p and zma-miR159k-3p genes were down-regulated in moderate stress conditions. The predicted targets of these miRNAs belong to the MYB-like transcription factor family, and a few MYB members have been demonstrated involvement in plant responses to biotic and abiotic stress conditions (Cao et al. 2013). In Arabidopsis, AtMYB96 mediated abscisic acid (ABA) signaling during drought stress (Seo et al. 2009), while AtMYB88 was able to control stomatal development during abiotic stress (Xie et al. 2010). Moreover, overexpression of different members of MYB-like transcription factor family improved drought stress tolerance in some species (Zhang et al. 2012; Cao et al. 2013; Tang et al. 2019). The antagonistic responses between miRNAs putatively regulating two different families of drought-related transcription factors (TFs MADS-box and MYB) during moderate stress in Setaria might be interesting to investigate, with hopes to establish the precise relationship between specific miRNA and its TF target.
During moderate drought stress, it was also observed that two miRNA genes were up-regulated, stu-miR408b-3p and tcc-miR530a, both possibly regulating cell wall-related genes (Supplementary File 6). The predicted target of stu-miR408b-3p refers to a laccase precursor protein (Sevir.9G440800), and the putative target of tcc-miR530a is an uncharacterized glycine-rich cell wall structural protein (Sevir.3G220100). Laccases are glycoproteins potentially involved in cell wall lignification, phenolic metabolism and H2O2 dynamics (Yu et al. 2017; Simões et al. 2020). Interestingly, the novel miRNA SvmiRp471 was also up-regulated in moderate stress and putatively targets a laccase gene (Sevir.8G221600), suggesting that laccases are down-regulated in early stages of moderate drought conditions. In sugarcane, ssp-miR397, a miRNA targeting a laccase gene, was up-regulated under early stages of drought stress, but it was down-regulated in late stages of dehydration, indicating a dynamic pattern of expression in stress conditions (Ferreira et al. 2012). In addition, several novel miRNA genes up-regulated in early responses of moderate stress appeared to target cell-wall related genes (Supplementary File 6), an indicative that cell wall signaling could be one of the first responses to dehydration.
miRNA-responsive genes during both moderate and severe drought stresses
The analysis of miRNA genes exclusively expressed in all stress conditions (moderate and severe), compared to well-irrigated plants, revealed two miRNA genes from the miR396 family (tcc-miR396d and zma-miR396f-5p) that were strongly down-regulated (Table 1; Supplementary File 6). It is known that miR396 controls the expression of differents growth-regulating factors (GRF), which are plant-specific transcription factors important for the maintenance of growth processes under unfavorable environmental conditions (Omidbakhshfard et al. 2015). In our in silico analysis, the predicted targets for tcc-miR396d and zma-miR396f-5p were observed as belonging to GRF family (Sevir.4G085500, Sevir.8G160800 and Sevir.9G140200; GO: 0,032,502). The miR396/GRF regulatory module controls a wide range of stress conditions, including high salinity and drought (Zhou et al. 2012; Liu et al. 2014; Omidbakhshfard et al. 2015), and the expression of these miRNA genes in early drought responses in S. viridis corroborates these previous studies.
miRNA-responsive genes during plant rehydration
After rehydration of plants, two miRNA genes that were strongly down-regulated in severe drought appeared to be up-regulated during recovery: osa-miR408-5p and zma-miR528b-3p (Fig. 4A; Table 1). The predicted targets of osa-miR408-5p are a stress responsive protein gene (SRP; Sevir.2G448800), coding for an unknown protein highly expressed in abiotic stress responses in maize (Vega-Arreguín et al. 2009), and a chalcone synthase gene (CHS; Sevir.7G023500), responsible for the entry point of the flavonoids pathway, representing enzymes that catalyze 4-coumaroy-CoA and malonyl-CoA to chalcone, leading the phenylpropanoids pathway to flavonoids biosynthesis (Shih et al. 2008). Flavonoid accumulation is likely to occur in regard to drought stress in several plant species (Castellarin et al. 2007; Yang et al. 2007; Ma et al. 2014) because they act as antioxidant in stressed plants (Ma et al. 2014) and overaccumulation of flavonoids improved drought tolerance in Arabidopsis and sugarcane (Wahid and Ghazanfar 2006; Nakabayashi et al. 2014). It is well established that one of the first responses to drought stress is the production of reactive oxygen species (ROS). Since antioxidant systems tightly regulate ROS signaling, even small changes in ROS generation or scavenging activity under drought stress are expected to have an instantaneous impact on signal transduction (de Carvalho 2008). In this context, one can speculate that in severe drought and subsequent rewatering the antioxidant system might be rapidly activated or deactivated, respectively, through the osa-miR408-5p/CHS module. However, we did not perform any experiment to confirm this hypothesis and further studies are necessary to address this question. The other miRNA gene that was inversely expressed in severe drought/rewatering, zma-miR528b-3p, putatively targets an uncharacterized gene (Sevir.5G176900) which encodes for a protein that demonstrated 40% identity to a FK506-binding protein (FKBPs) from Panicum hallii. These proteins appeared to be chaperones playing various functions in a variety of crucial processes for plant development, growth, and abiotic stress responses (Gollan et al. 2012; Dong et al. 2018) and further investigation of the zma-miR528b-3p/ Sevir.5G176900 module could reveal its function and its role in drought stress responses.
Principal components analysis
The data for the different levels of drought stress from physiological analyses, in silico expression and RT- qPCR for the validated miRNAs was submitted to principal components analysis (PCA). For the SvmiRp86, miR159a and zma-miR396f-5p it was possible to distinguish in two different ellipses the expression of miR and their targets (Supplementary Fig. S3). In the brown ellipse the miR in silico expression in green symbols and miR RT-qPCR expression in blue symbols grouped and mostly modulated for the physiological parameters Gs, E and WUE in the several stress conditions (25% and 0% drought stress). The respective targets of these miRs in the different drought levels were grouped into a gray ellipse, and were not included for the physiological parameters analyzed (Supplementary Fig. S3).
Comparison of miRNA regulation in different millet species during drought stress
Setaria viridis, also known as green millet, is a diploid C4 grass member of subfamily Panicoideae, tribe Paniceae, that is emerging as a model organism due the short life cycle, small stature, large seed yield, and a small genome (510 Mb). Thus, green millet is a interesting model for research molecular mechanisms of C4 photosynthesis, as well as, validate biotechnological techniques for increase plant yields in phylogenetically related crops, such as sorghum, pearl millet, maize, and sugarcane (Cesarino et al. 2020).
Wang et al. (2016) performed small RNA and degradome sequencing to discover miRNAs and their targets in foxtail millet (Setaria italica) during drought responses. The genomes of S. italica and S. viridis are closely related (~ 99%), and it is tempting to assume that miRNA responses to drought stress would be similar between these two species. In fact, several miRNAs were responsive in a similar way in green and foxtail millets during drought stress. The families of conserved miRNAs belonging to miR167, 398, 159, 396, 408, 171, 395, 827, 528 and 164 were all differentially expressed in a similar pattern in both species in response to drought. For instance, miR164 was upregulated during drought stress in both species, and its predicted target corresponds to no apical meristem protein transcription factor (NAC). As discussed by Wang et al. (2016), it is believed that the increase in the expression of miR164 reduce the NAC gene expression and regulate the foxtail millet responds to drought, and our results contribute to reinforce this hypothesis in green millet. In addition, miR159 was differentially regulated in drought stress in both studies, and their predicted targets comprise MYB transcription factors, responsible for the regulation of ABA signaling. The similar responses obtained in S. italica and S. viridis corroborate the idea that green millet is a suitable model for genetic studies in C4 grasses, especially millets.
Moreover, Chakraborty et al. (2020) chosen five important millet species (pearl millet, sorghum, foxtail millet, finger millet, and proso millet) to mine stress-responsive miRNAs using in silico analysis. The authors demonstrated that many of the miRNA families targeting drought-responsive factors were both functionally and structurally conserved between the species, showing that millets regulatory roles remained largely conserved. Several miRNA families associated with drought-responsive traits were identified in foxtail millet that are in accordance with our study in green millet, namely miR105, 171, 164, 395, 167, 319, 172, 165 and 399 (Chakraborty et al. 2020; this study, Table 1). Therefore, our results indicate that these miRNA families and their targets associated with drought stress are indeed conserved among millet species. However, it should be addressed that other miRNA families were exclusively expressed in green millet under dehydration.
Regulation of cell wall-related genes by novel miRNAs during early responses to drought
Our in silico analysis in green millet also reveals that several cell wall-related genes were predicted to be regulated by novel miRNAs differentially expressed in early responses of drought stress in Setaria viridis (Supplemenary File 6). Cell wall synthesis and remodeling is a mechanism employed by plants under abiotic stress, mainly because the major issue during a drought is to restrict the shoot development and sustain the root growth (Tenhaken 2015; Le Gall et al. 2015; Oliveira et al. 2020). As mentioned above, the production of ROS and peroxidases is a primary response to biotic and abiotic stresses (de Carvalho 2008), which are responsible for the cross-linking of phenolic compounds and glycoproteins in the cell walls, causing stiffening. Moreover, cell wall-related enzymes are also responsible to modulate cell growth through the modification of the cell wall matrix, resulting in wall loosening to allow further growth of stressed organs (Tenhaken et al. 2015). Our results demonstrated that seven novel miRNA genes up-regulated during drought stress (SvmiRp86, 453, 466, 471, 730, 751 and 826) are predicted to target a wide range of cell wall-related genes coding for UDP-glucosyl transferase, laccase, β-mannanase, glycosil transferase, glycosil hydrolase, xyloglucan fucosyltransferase and pectin methylesterase (Supplementary File 6). A single novel miRNA, SvmiRp86, was down-regulated in severe stress, and this miRNA gene was predicted to target different cell wall genes belonging to the families of expansins, glycosil hydrolases 16, β-glucosidases and α-1,4-glucanases. All these cell wall-modifying genes had their expression negatively correlated with SvmiRp86 expression, as corroborated by RT-qPCR data (Fig. 4B; Supplementary Fig. 2). Altogether, these results indicate a strict regulation of cell wall during early responses of drought stress and recovery of Setaria plants. Therefore, it would be interesting to perform detailed analyses on the relationship between the miRNA, their targets and cell wall structure in plants under abiotic stress and during recovery from drought.
Conclusions
In conclusion, our results demonstrated a fine-tuning of miRNA regulation during early responses of Setaria viridis plants to drought and after recovery. The predicted mRNA targets mainly corresponded to genes related to cell wall remodeling, growth, antioxidant system and drought-related transcription factors. Ecophysiological studies demonstrated that the S. viridis accession used in this study is highly tolerant to drought and capable of a rapid recovery response, demonstrating an adaptive mechanism to cope with abiotic stress. Therefore, the genetic engineering of miRNAs and their predicted targets represents a promising strategy to improve drought stress tolerance in crops phylogenetically related to 1`S. viridis, such as sugarcane, maize, and sorghum.
Data availability
The data were availability in https://www.ncbi.nlm.nih.gov/genbank/ and the accession numbers could be found in the supplementary file 3.
References
Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ (2008) Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 18:758–762
Addo-Quaye C, Miller W, Axtell MJ (2009) CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25:130–131
Arora R, Agarwal P, Ray S, Singh AK, Singh VP, Tyagi AK, Kapoor S (2007) MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8:242
Aydinoglu F (2020) Elucidating the regulatory roles of microRNAs in maize (Zea mays L.) leaf growth response to chilling stress. Planta 251:1–15
Barciszewska-Pacak M, Milanowska K, Knop K et al (2015) Arabidopsis microRNA expression regulation in a wide range of abiotic stress responses. Front Plant Sci 6:410
Basso MF, Ferreira PCG, Kobayashi AK et al (2019) MicroRNAs and new biotechnological tools for its modulation and improving stress tolerance in plants. Plant Biotechnol J 17:1482–1500
Bennetzen JL, Schmutz J, Wang H, Percifield R et al (2012) Reference genome sequence of the model plant Setaria. Nat Biotechnol 30:555–561
Borges F, Martienssen RA (2015) The expanding world of small RNAs in plants. Nat Rev Mol Cell Biol 16:727–741
Brutnell TP, Wang L, Swartwood K, Goldschmidt A et al (2010) Setaria viridis: a model for C4 photosynthesis. Plant Cell 22:2537–2544
Brutnell TP, Bennetzen JL, Vogel JP (2015) Brachypodium distachyon and Setaria viridis: Model Genetic Systems for the Grasses. Ann Review Plant Biol 66:465–485
Burney J, Ramanathan V (2014) Recent climate and air pollution impacts on Indian agriculture. PNAS 111:16319–16324
Cao ZH, Zhang SZ, Wang RK, Zhang RF, Hao YJ (2013) Genome wide analysis of the apple MYB transcription factor family allows the identification of MdoMYB121 gene conferring abiotic stress tolerance in plants. PLoS ONE 8:e69955
Carvalho De (2008) Drought stress and reactive oxygen species. Plant Signal Behav 3(3):156–165
Castellarin SD, Matthews MA, Di Gaspero G, Gambetta GA (2007) Water deficits accelerate ripening and induce changes in gene expression regulating flavonoid biosynthesis in grape berries. Planta 227:101–112
Cesarino I, Dello Ioio R, Kirschner GK et al (2020) Plant science’s next top models. Ann Bot 126(1):1–23
Chakraborty A, Viswanath A, Malipatil R et al (2020) Structural and functional characteristics of miRNAs in five strategic millet species and their utility in drought tolerance. Front Genet 11:608421
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH et al (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33:e179
De Souza WR, Martins PK, Freeman J, Pellny TK et al (2018) Suppression of a single BAHD gene in Setaria viridis causes large, stable decreases in cell wall feruloylation and increases biomass digestibility. New Phytol 218:81–93
De Souza WR, Pacheco TF, Duarte KE, Sampaio BL et al (2019) Silencing of a BAHD acyltransferase in sugarcane increases biomass digestibility. Biotechnol Biofuels 12:111
Dong Q, Mao K, Duan D, Zhao S et al (2018) Genome-wide analyses of genes encoding FK506-binding proteins reveal their involvement in abiotic stress responses in apple. BMC Genomics 19:707
Ermawar RA, Collins HM, Byrt CS et al (2015) Genetics and physiology of cell wall polysaccharides in the model C4 grass, Setaria viridis spp. BMC Plant Biol 15:1–18
Farooq M, Wahid A, Kobayashi N, Fujita D, Basra SMA (2009) Plant drought stress: effects, mechanisms and management. In: Lichtfouse E, Navarrete M, Debaeke P, Véronique S, Alberola C (eds) Sustainable Agriculture. Springer, Netherlands, pp 153–188
Ferdous J, Hussain SS, Shi B-J (2015a) Role of microRNAs in plant drought tolerance. Plant Biotechnol J 13:293–305
Ferdous J, Li Y, Reid N, Langridge P, Shi BJ, Tricker PJ (2015b) Identification of reference genes for quantitative expression analysis of microRNAs and mRNAs in barley under various stress conditions. PLoS ONE 10:e0118503
Ferreira TH, Gentile A, Vilela RD et al (2012) microRNAs Associated with Drought Response in the Bioenergy Crop Sugarcane (Saccharum spp.). PLoS ONE 7(10):e46703
Frosi G, Harand W, de Oliveira MT et al (2017) Different physiological responses under drought stress result in different recovery abilities of two tropical woody evergreen species. Acta Botanica Brasilica 31(2):153–160
Gao R, Wang Y, Gruber MY, Hannoufa A (2018) miR156/SPL10 modulates lateral root development, branching and leaf morphology in arabidopsis by silencing AGAMOUS-LIKE 79. Front Plant Sci 8:2226
Geng Z, Liu J, Li D et al (2021) A Conserved miR394-targeted F-Box gene positively regulates drought resistance in Foxtail Millet. J Plant Biol 64:243–252
Gollan PJ, Bhave M, Aro EM (2012) The FKBP families of higher plants: Exploring the structures and functionsof protein interaction specialists. FEBS Lett 586(20):3539–3547
Guerra D, Crosatti C, Khoshro HH et al (2015) Post-transcriptional and post-translational regulations of drought and heat response in plants: a spider’s web of mechanisms. Front Plant Sci 6:57
Hackenberg M, Gustafson P, Langridge P, Shi B-J (2015) Differential expression of microRNAs and other small RNAs in barley between water and drought conditions. Plant Biotechnol J 13:2–13
Hajyzadeh M, Turktas M, Khawar KM, Unver T (2015) miR408 overexpression causes increased drought tolerance in chickpea. Gene 555:186–193
Huang P, Brutnell TP (2016) A synthesis of transcriptomic surveys to dissect the genetic basis of C4 photosynthesis. Curr Opin Plant Biol 31:91–99
Jeong DH, Park S, Zhai J, Gurazada S et al (2011) Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 23:4185–4207
Kim JH, Woo HR, Kim J, Lim PO et al (2009) Trifurcate feed-forward regulation of age-dependent cell death involving miR164 in Arabidopsis. Science 323:1053–1057
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68-73
Kozomara A, Birgaoanu M, Griffiths-Jones S (2019) miRBase: from microRNA sequences to function. Nucleic Acids Res 47:D155–D162
Lawrence D, Vandecar K (2015) Effects of tropical deforestation on climate and agriculture. Nature 5:27–36
Le Gall H, Philippe F, Domon JM, Gillet Fm Pelloux J, Rayon C (2015) Cell Wall Metabolism in Response to Abiotic Stress. Plants 4:112–166
Li P, Brutnell TP (2011) Setaria viridis and Setaria italica, model genetic systems for the Panicoid grasses. J Exp Bot 62:3031–3037
Liu H, Guo S, Xu Y, Li C, Zhang Z, Zhang D et al (2014) OsmiR396d-regulated OsGRFs function in floral organogenesis in rice through Binding to Their Targets OsJMJ706 and OsCR4. Plant Physiol 165(1):160–174
Liu T, Fang C, Ma Y, Shen Y, Li C, Li Q et al (2016) Global investigation of the co-evolution of MIRNA genes and microRNA targets during soybean domestication. Plant J 85:396–409
Lu C, Meyers BC, Green PJ (2007) Construction of small RNA cDNA libraries for deep sequencing. Methods 43:110–117
Ma D, Sun D, Wang C, Li Y, Guo T (2014) Expression of flavonoid biosynthesis genes and accumulation of flavonoid in wheat leaves in response to drought stress. Plant Phys Biochem 80:60–66
Martins PK, Mafra V, de Souza WR, Ribeiro AP et al (2016) Selection of reliable reference genes for RT-qPCR analysis during developmental stages and abiotic stress in Setaria viridis. Sci Rep 6:28348
Matzke MA, Mosher RA (2014) RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nature 15:394–408
Matzke MA, Kanno T, Matzke AJ (2015) RNA-directed DNA Methylation: the evolution of a complex epigenetic pathway in flowering plants. Ann Rev Plant Biol 66:243–267
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Nakabayashi R, Yonekura-Sakakibara K, Urano K et al (2014) Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of antioxidant flavonoids. Plant J 77:367–379
Ni Z, Hu Z, Jiang Q, Zhang H (2012) Overexpression of gma-MIR394a confers tolerance to drought in transgenic Arabidopsis thaliana. Biochem Biophys Res Commun 427:330–335
Oliveira DM, Mota TR, Salatta FV, Sinzker RC et al (2020) Cell wall remodeling under salt stress: Insights into changes in polysaccharides, feruloylation, lignification, and phenolic metabolism in maize. Plant Cell Environ 43:2172–2191
Omidbakhshfard MA, Proost S, Fujikura U, Mueller-Roeber B (2015) Growth-Regulating Factors (GRFs): a small transcription factor family with important functions in plant biology. Mol Plant 8:998–1010
Pegler JL, Duc QN, Christopher PL et al (2020) Profiling of the salt stress responsive microRNA landscape of C4 genetic model species Setaria viridis (L.) Beauv. Agronomy 10(6):837
Puig J, Meynard D, Khong GN, Pauluzzi G et al (2013) Analysis of the expression of the AGL17-like clade of MADS-box transcription factors in rice. Gene Expr Patterns 13:160–170
Ribeiro AP, de Souza WR, Martins PK, Vinecky F et al (2017) Overexpression of BdMATE gene improves aluminum tolerance in Setaria viridis. Front Plant Sci 8:1–12
Rosenzweig C, Elliott J, Deryng D, Ruane AC et al (2014) Assessing agricultural risks of climate change in the 21st century in a global gridded crop model intercomparison. Proc Natl Acad Sci 111:3268–3273
Saha P, Sade N, Arzani A, Wilhelmi MMR et al (2016) Effects of abiotic stress on physiological plasticity and water use of Setaria viridis (L.). Plant Sci 251:128–138
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101
Seo PJ, Xiang F, Qiao M, Park JY, Lee YN, Kim SG et al (2009) The MYB96 transcription factor mediates abscisic acid signaling during drought stress response in Arabidopsis. Plant Physiol 151(1):275–289
Shih CH, Chu H, Tang LK, Sakamoto W et al (2008) Functional characterization of key structural genes in rice flavonoid biosynthesis. Planta 228:1043–1054
Simões MS, Carvalho GG, Ferreira SS et al (2020) Genome-wide characterization of the laccase gene family in Setaria viridis reveals members potentially involved in lignification. Planta 251:46
Song JB, Gao S, Sun D, Li H, Shu XX, Yang ZM (2013) miR394 and LCR are involved in Arabidopsis salt and drought stress responses in an abscisic acid-dependent manner. BMC Plant Biol 13:1–16
Stocks MB, Moxon S, Mapleson D, Woolfenden HC et al (2012) The UEA sRNA workbench: a suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 28:2059–2061
Tang Y, Bao X, Zhi Y, Wu Q, Guo Y et al (2019) Overexpression of a MYB family gene, OsMYB6, increases drought and salinity stress tolerance in transgenic rice. Front Plant Sci 10:168
Tenhaken R (2015) Cell wall remodeling under abiotic stress. Front Plant Sci 5:771
Varkonyi-Gasic E, Wu R, Wood M et al (2007) Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3:12
Vega-Arreguín JC, Ibarra-Laclette E, Jiménez-Moraila B et al (2009) Deep sampling of the Palomero maize transcriptome by a high throughput strategy of pyrosequencing. BMC Genomics 10:299
Wahid A, Ghazanfar A (2006) Possible involvement of some secondary metabolites in salt tolerance of sugarcane. J Plant Physiol 163(7):723–730
Wang Z, Hu H, Goertzen LR, McElroy JS, Dane F (2014) Analysis of the Citrullus colocynthis transcriptome during water deficit stress. PLoS ONE 9(8):e104657
Wang M, Li A-M, Liao F et al (2022) Control of sucrose accumulation in sugarcane (Saccharum spp. hybrids) involves miRNA-mediated regulation of genes and transcription factors associated with sugar metabolism. GCB Bioenergy 14:173–191
Xie Z, Li D, Wang L, Sack FD, Grotewold E (2010) Role of the stomatal development regulators FLP/MYB88 in abiotic stress response. Plant J 64:731–739
Yang Y, He F, Yu L, Chen X, Lei J, Ji J (2007) Influence of drought on oxidative stress and flavonoid production in cell suspension culture of glycyrrhiza inflata Batal. Zeitschrift Für Naturforschung C 62:410–416
You Q, Zhang L, Yi X, Zhang Z, Xu W, Su Z (2015) SIFGD: setaria italica functional genomics database. Mol Plant 8:967–970
Yu Y, Li Q-F, Zhang J-P, Zhang F et al (2017) Laccase-13 regulates seed setting rate by affecting hydrogen peroxide dynamics and mitochondrial integrity in rice. Front Plant Sci 8:1324
Yue E, Liu Z, Li C et al (2017) Overexpression of miR529a confers enhanced resistance to oxidative stress in rice (Oryza sativa L.). Plant Cell Rep 36(7):1171–1182
Zhang L, Zhao G, Xia C, Jia J, Liu X, Kong X (2012) A wheat R2R3-MYB gene, TaMYB30-B, improves drought stress tolerance in transgenic Arabidopsis. J Exp Bot 63(16):5873–5885
Zhou J, Liu M, Jiang J et al (2012) Expression profile of miRNAs in Populus cathayana L. and Salix matsudana Koidz under salt stress. Mol Biol Rep 39:8645–8654
Zhou M, Li D, Li Z et al (2013) Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic creeping bentgrass. Plant Physiol 161:1375–1391
Acknowledgements
The authors are grateful to Dirk Winkelman to language editing.
Funding
MFB is grateful to CNPq for a postdoctoral research fellowship (PDJ 150936/2018–4). HBCM is grateful to FAP-DF from Edital Universal 03/2016 and EMBRAPA from Macroprograma 03/2017 and 02/2017 (02.16.05.022.00.00). WRS (2019/04878–7) and KED (2019/16226–4) are grateful to FAPESP for the financial support.
Author information
Authors and Affiliations
Contributions
MFB, NGO, WRS and HBCM designed all the experiments; MFB, KED and NGO conducted all biological experiments; MFB, JCFS and TBC performed all bioinformatics analysis; MFB, KED, BADBC, BOG and TRS carried out molecular assays (real time RT-PCR and Stem-loop RT-PCR); MFB wrote the draft manuscript, while WRS, ALN and AKK provided intellectual inputs. WRS wrote the final version of the manuscript. All authors revised and approved the final version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Duarte, K.E., Basso, M.F., de Oliveira, N.G. et al. MicroRNAs expression profiles in early responses to different levels of water deficit in Setaria viridis. Physiol Mol Biol Plants 28, 1607–1624 (2022). https://doi.org/10.1007/s12298-022-01226-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-022-01226-z