
Abstract
Rheum tanguticum (Maxim. ex Regel) Maxim. ex Balf. is a herbaceous perennial plant indigenous to China, and its root and rhizomes were usually used as an important traditional Chinese medicine. However, the genomic resources are still scarce for R. tanguticum and even for Rheum genus. Transcriptome datasets from different tissues of R. tanguticum were obtained to screen the genes related to anthraquinones biosynthesis, and five free anthraquinones were also determined. Nine cDNA libraries of roots, stems and leaves were generated, and a total of 272 million high-quality reads were assembled into 257,942 unigenes. Based on the functional annotation, A total of 227 candidate enzyme genes involved in the MVA, MEP, shikimate and polyketide pathways were identified, and several differentially expressed genes found functionally associated with anthraquinones biosynthesis showed distinct tissue-specific expression patterns. Especially, we found that the expression levels of PKS III genes might result in the content differences of free anthraquinones in different tissues of R. tanguticum. Besides, 137,400 SSR loci were identified, and 64,081 SSR primer pairs were successfully designed based on these loci. Our results not only provide cues for the genetic mechanism of anthraquinone content differences in different tissues of R. tanguticum, but also lay genomic foundation for the subsequent genetic engineering and breeding for Rheum species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rheum tanguticum (Maxim. ex Regel) Maxim. ex Balf. is one of the source plants of rhubarb that prefers to growing in the mountainous areas with a high altitude. The dried roots of R. tanguticum is predominantly valued for its medicinal uses as an important traditional Chinese medicine. There are plethora of studies focusing on the pharmacodynamic effects of rhubarb and indicating that the dominant chemical constituents of rhubarb are anthraquinones and their derivatives (Aichner and Ganzera 2015; Danielsen et al. 1992; Koyama et al. 2007). It has been proven that anthraquinones and their derivatives harbor various pharmacological activities such as detoxification, removal of blood stasis, removing dampness, abating jaundice, etc. (Chinese Pharmacopoeia Committee 2015; Zhou et al. 2020). As rhubarb has important value and great market demand, the resource reserve of R. tanguticum in wild populations has seriously declined and even faced extinction in several areas (Li et al. 2014). Recently, artificial cultivation regeneration systems have been developed, the chemical diversity and genetic variation of rhubarb populations has been preliminarily elucidated (Ren et al. 2016; Wang et al. 2018). However, there are still limited research on the functional genomics of Rheum species, and the biosynthesis pathways of active ingredients are also not well clarified. Therefore, it is necessary to explore more genomic resources which may lay a genetic foundation for the molecular marker-assisted breeding of rhubarb and the promotion of production for its active constituents.
In higher plants, anthraquinones are mainly derived from two main biosynthetic pathways: the polyketide pathway and a combination of shikimate and mevalonate (MVA)/methyl-D-erythritol 4-phosphate (MEP) pathways (Han et al. 2001; Kang et al. 2020) (Fig. 1). In polyketide pathway, an octaketide chain was produced by the combination of seven molecules of malonyl-CoA and an acetyl-CoA unit, and these reactions are catalyzed by type III polyketide synthase (PKS) enzymes (Karppinen et al. 2008; Mizuuchi et al. 2009). Subsequently, the core unit of polyketides, which are the scaffolds for the formation of anthraquinones, were produced from the linear octaketide intermediate by a series of cyclization and decarboxylation reactions (Karppinen et al. 2008; Mizuuchi et al. 2009; Pillai and Nair 2014; Yamazaki et al. 2013). In the shikimate pathway, the rings A and B of anthraquinones are originated from 1,4-dihydroxy-2-naphthoyl-CoA which is synthesized by isochorismate and α-ketoglutarate, whereas ring C of anthraquinones is derived from 1,4-dihydroxy-2-naphthoyl-CoA via the MVA/MEP pathway (Han et al. 2001; Rama Reddy et al. 2015) (Fig. 1). In addition, Cyt P450s (CYPs) may play important roles in some post-strictosamide steps for the modifications of the backbone of anthraquinones, and UDP-glycosyltransferases (UGTs) have been proven to be related to the biosynthesis of anthraquinones glycosides (Rama Reddy et al. 2015) (Fig. 1). However, no comprehensive genetics or molecular studies of anthraquinones biosynthesis were conducted in R. tanguticum. Therefore, whether the abovementioned biosynthetic pathways and the related key enzymes were available for R. tanguticum is still unclear.

Putative pathway for anthraquinones biosynthesis. The enzyme names with red color indicate that their candidate coding genes are detected in this study
Deep transcriptome sequencing (RNA-seq) for medicinal plants provided an opportunity to discover genes involved in the biosynthetic pathways that lead to the biosynthesis of secondary metabolites in plant species (Yamazaki et al. 2013). Moreover, transcriptomic data can provide insights into gene regulation and expression changes in complex networks and facilitate the identification of polymorphism markers that are mostly within or around gene-encoding regions (Zhou et al. 2016a, b). Transcriptomics-enabled gene discovery strategies have been adopted in many medicinal plants to identify candidate structural genes involved in the biosynthesis of secondary metabolites such as flavonoids, terpenoid and alkaloid (Amini et al. 2019; Shen et al. 2017; Yuan et al. 2020). Several structural genes involved in anthraquinone biosynthesis have been identified for Rheum officinale Baill. and Rheum palmatum Linn. based on the transcriptome sequencing (Hei et al. 2019; Li et al. 2018), and another study further indicated that some structural genes were differentially expressed between these two Rheum species (Liu et al. 2020). However, aforementioned studies only identified the anthraquinone-related genes using the transcriptomic data derived from seedlings or fresh leaves, which may not provide comprehensive genetic information due to the restriction of temporality and spatiality of transcriptome. It has been reported that anthraquinone contents have large differences in different tissues of Rheum species (Li et al. 2019). But no research was implemented to explore the reasons of these differences based on specific structural gene expression levels, and the expression profiling of structural gene involved in anthraquinone biosynthesis in specific tissues of Rheum species still remains unknown.
In the present study, we conducted the comparative transcriptome analyses of the roots, stems and leaves of R. tanguticum. We intended to identify the key genes involved in the biosynthesis and regulation of anthraquinones based on transcriptomic data of R. tanguticum and examine the gene expression levels in different tissues. Our results will not only enhance the genomic foundation for the following genetic engineering and breeding for rhubarb, but also provide more cues for the genetic mechanism of anthraquinone content differences in different tissues.
Materials and methods
Plant materials
Rheum tanguticum was collected from Zeku county, Qinghai province, China (35°15′ N, 101°53′ E). Roots, stems, and leaves from the same individual at flowering age were collected, and three individuals were simultaneously sampled as biological replicates. Total nine samples were harvested in liquid nitrogen immediately and stored at − 80 °C.
The measurement of the content of five free anthraquinones
Root, stem and leaf tissues of R. tanguticum were powdered using a mixer mill (MM 400, Ningbo Scientz Biotechnology Co., Ltd., Ningbo, China) with a zirconia bead for 1.5 min at 30 Hz. All the procedures for extraction and HPLC measurements in the present study were described in Chinese Pharmacopoeia (Chinese Pharmacopoeia Committee 2015). Briefly, 500 mg sample of the powder was weighed and mixed with 25 mL of 100% methanol solvent (v/v) in a screwed glass vial (12 × 2 cm) and placed in boiling water bath (65 °C) for 60 min. The residue was dissolved in methanol and transferred to a 25-mL volumetric flask for the subsequent HPLC measurements. Three biological replicates were used to ensure the reliability. HPLC analysis was performed using an Agilent 1100 system (Agilent Technologies, Palo Alto, CA). Chromatographic analysis was conducted using a Sepax RP-C18 column (Amethyst C18-H, 4.6 mm × 250 mm, particle size: 5 μm; Sepax Technologies, Suzhou, China) maintained at 30 °C. The mobile phase consisted of 0.1% (v/v) aqueous phosphoric acid and methanol. The detection wavelength was 254 nm. The injected volume was 10 μL, and velocity of flow was set as 1 mL/min. The validations of HPLC method for precision, accuracy, specificity, system suitability, peak purity and robustness were conducted using the procedures in our previous study (Zhu et al. 2021). The content of five free anthraquinones was calculated using standard graphs of aloe-emodin, rhein, emodin, chrysophanol and physcion (mg g−1). ANOVA one-way analysis of variance was conducted with GraphPad Prism 9 (www.graphpad.com) to detect differences in five free anthraquinones between the three different tissues of R. tanguticum.
RNA extraction, cDNA library construction and Illumina sequencing
Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA). RNA concentration was evaluated using a Qubit 2.0 fluorometer (Life Technologies, CA), and RNA quality was verified with 2% agarose gels. RNA purity was checked using a NanoDrop ND-2000 spectrophotometer (NanoDrop products, Wilmington, DE, USA). RNA integrity was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The cDNA libraries were constructed using a previously published procedures (Zhou et al. 2016a, b) and then sequenced on the Illumina X Ten platform with the sequence length of 150 bp.
De novo transcriptome assembly and functional annotation
Raw reads derived from Illumina sequencing were first trimmed to obtain high quality reads by removing adaptor sequences, reads with unknown base calls (N) more than 5%, and low-quality reads. After trimming, the clean reads were assembled to be transcripts using assembler Trinity v2.5.1 with the default parameters (Grabherr et al. 2011). Afterwards, the assembled transcripts were further processed by CD-HIT v4.6 with a sequence identity threshold of 0.95 to remove redundancies (Fu et al. 2012), and the resultant unigenes were used for following steps. In order to know the functions of transcriptome sequences, all non-redundant unigenes were searched against the public databases, including NCBI non-redundant protein (nr), Swiss-Prot, Cluster of Orthologous Group (COG), euKaryotic Ortholog Group (KOG) and Translation of EMBL (TrEMBL) using BLASTX with an E-value threshold of 1E-5 (Altschul et al. 1997). Kyoto Encyclopedia of Genes and Genomes (KEGG) classification was performed using the KEGG Automatic Annotation Server (KAAS) with an E-value of 1E-10. Protein family (Pfam) alignments were carried out using the HMMER v3.0 (http://hmmer.org/) with an E-value of 1E-5. Gene ontology (GO) analysis for whole assembled transcriptome was performed based on the annotation results of nr database using Blast2GO v2.5 with an E-value of 1E-5 (Conesa et al. 2005).
Genes related to type III polyketide synthases in R. tanguticum and phylogenetic analyses
In order to identify candidate genes encoded type III polyketide synthases (PKS), we downloaded 138 protein sequences belongs to type III PKS from GenBank (accession numbers were summarized in Table S1). Some of these sequences were classified as the chalcone synthases (CHS) superfamily of type III PKS. Unigene sequences of this study was retrieved to predict open reading frames (ORF) by the Getorf program with a minimum length of 150 amino acids (Rice et al. 2000), and then 138 protein sequences were searched against the ORFs of unigenes using BLASTP with an E-value threshold of 1E-5 (Altschul et al. 1997). After alignments, 8 amino acid sequences of ORF-predicted unigenes showed high similarity to the type III PKS were identified and used for the following analyses. For inferring the phylogeny of type III PKS family in Polygonaceae, 50 protein sequences belong to type III PKS from GenBank (Table S1) and 8 protein sequences from this study were retrieved for phylogenetic inference with the bacterial type III PKS Mycobacterium tuberculosis PKS18 was set as an outgroup. All the protein sequences were aligned with MUSCLE algorithm in MEGA X (Kumar et al. 2018). The ML phylogenetic tree was constructed using IQ-TREE v2.1.2 (Minh et al. 2020) with the best-fit model selected by ModelFinder (Kalyaanamoorthy et al. 2017), and the bootstrap replicates were set as 1000 steps.
Tissue-specific differential expression analysis
The differential expression analysis was performed to determine the gene expression from target processes in different tissues. The RSEM v1.2.29 with default parameters was used to estimate the gene expression level of each sample based on FPKM (Fragments Per Kilobase of transcript per Million mapped reads) (Li and Dewey 2011). Differentially expressed genes (DEGs) between each two samples were identified using DESeq2 R package (Anders and Huber 2010). Fold-change values between samples were estimated based on the FPKM values. The |log2(fold change)|> 1, and false discovery rate (FDR) < 0.05 was set as the threshold to assess the significance level of differential gene expression. The Venn diagram of DEGs derived from pairwise comparison of different organs was conducted using ClusterVenn (Xu et al. 2019). In order to determine the potential functions and metabolic pathways of these DEGs, all the DEGs were also searched against the nr, Swiss-Prot, GO, COG, KOG, TrEMBL, Pfam, and KEGG databases. ClusterProfile v3.14.0 (Yu et al. 2012) was used to perform the statistical enrichment of DEGs in GO and KEGG pathways.
Quantitative real-time PCR (qRT-PCR) analysis
In order to validate the RNA-seq results, 14 DEGs related to anthraquinones synthesis were selected for the qRT-PCR validation using the specific primers designed by Primer 5. The housekeeping gene Actin and Hist were selected as the internal control for normalization. All the primers are shown in Table S2. Total RNA of each sample was extracted using the aforementioned procedures. After the removal of genomic DNA, the cDNA synthesis was conducted using NovoScript® one-step 1st Strand cDNA Synthesis SuperMix (Novoprotein, Shanghai, China). The qRT-PCR was performed using NovoStart® SYBR qPCR SuperMix Plus (Novoprotein, Shanghai, China). All qRT-PCR reactions were performed in BIO-RAD CFX Connect Real-Time PCR Detection System (BIORAD, USA) as follow: 95 °C for 60 s, followed by 40 cycles of 95 °C for 5 s, 60 °C for 10 s, and at 72 °C for 15 s. All samples were subjected to three technical replicate experiments to ensure the reliability. Finally, the relative gene expression was calculated based on the 2−ΔΔCt method (Schmittgen and Livak 2008).
Identification of simple sequence repeat (SSR) loci
All the non-redundant unigenes from R. tanguticum transcriptome were used to identify the candidate SSR loci. Microsatellite screening was conducted using MISA perl script (Thiel et al. 2003) with parameters for identifying SSR as six for di-, four for tri-, three for tetra- and penta-, and two for hexa-nucleotide motifs, respectively. Mono-nucleotide repeats were excluded in our analyses due to base mismatch or sequencing errors. The SSRs with the suitable flanking lengths were selected to design the PCR primers using the program Primer 3 (Rozen and Skaletsky 1999). The criteria for designing primers were as follows: PCR product size range of 100 to 300 bp; primer length of 18–23 nucleotides; GC content of 30–70% and annealing temperature between 50–70 °C with 55 °C as the optimum melting temperature. Finally, 38 primer pairs (Table S3) were randomly selected for amplification using DNA from the Rheum species (R. officinale, R. tanguticum, R. tanguticum var. liupanshanense, R. palmatum). PCR amplifications were conducted in a reaction volume of 10 µL with 5 µL 2 × Taq PCR Master Mix, 0.2 µM of each primer, 1 µL template DNA and 3.6 µL ddH2O. All amplifications were carried out in SimpliAmp™ Thermal Cycler (Applied Biosystems, Carlsbad, CA, USA) as follow: denaturation at 94 °C for 5 min, followed by 30 cycles of 94 °C for 50 s, at specific annealing temperature (Tm) for 30 s, 72 °C for 40 s and 72 °C for 5 min as final extension. PCR products were visualized on 2% agarose gels after staining with ethidium bromide.
Results
The content of five free anthraquinones
Five free anthraquinones including aloe-emodin, rhein, emodin, chrysophanol and physcion were determined in roots (Rt_R), leaves (Rt_L) and stems (Rt_S) of R. tanguticum, respectively. The content of aloe-emodin, rhein, emodin, chrysophanol and physcion was 2.46 mg/g, 4.35 mg/g, 0.88 mg/g, 4.00 mg/g and 0.88 mg/g in roots, respectively. The content was 0.35 mg/g, 0.24 mg/g, 0.94 mg/g, 0.70 mg/g and 0.10 mg/g in leaves and was 0.05 mg/g, 0.06 mg/g, 0.16 mg/g, 0.52 mg/g and 0.01 mg/g in stems, respectively (Fig. 2). Our results indicated that the highest content of free anthraquinone was detected in the roots of R. tanguticum, and the stems shared lowest content of free anthraquinone except for emodin. ANOVA one-way analysis of variance showed that there were significant differences in the content of emodin between the three different tissues of R. tanguticum. The content differences for other anthraquinones (Aloe-emodin, Chrysophanol, Physcion, Rhein) were not significant between stems and leaves but they were significant between roots and other two tissues (Fig. 2, Table S4).

The content of five free anthraquinones in different tissues. Lowercase letters represent significant differences between different tissues
Summary statistics of RNA-seq
Nine cDNA libraries were prepared and used for the RNA-seq on the Illumina X Ten platform, and the libraries were named as follows: cDNA libraries of root: Rt_R1, Rt_R2, Rt_R3; cDNA libraries of leaf: Rt_L1, Rt_L2; Rt_L3; cDNA libraries of stem: Rt_S1, Rt_S2, Rt_S3. After sequencing and trimming, clean reads generated from each library ranged from 27,280,046 to 39,364,810 (Table S5). The Q30 value of each sample was up to 93.69%, and the GC content of each sample ranged from 49.16 to 50.65% (Table S5). The results showed that these high-quality reads could be used for following analysis.
De novo assembly and functional annotation
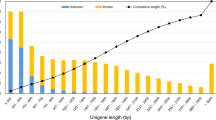
The clean reads derived from nine cDNA libraries of three different tissues were pooled and assembled according to the de novo assembly strategy by Trinity. After assembly, a total of 6,26,482 transcripts were recovered with an average length of 1387.47 bp and N50 of 2485 bp. And then the resultant transcripts were further used to remove the redundancy. Finally, 2,57,942 unigenes with an average length of 573.90 bp and N50 of 834 bp were recovered for the subsequent functional annotation and DEG analyses (Table 1). The length distribution of the unigenes was shown in Fig. S1.
According to the similarity searching against to eight databases, approximately 57.51% of the unigenes (148,344) had at least one blast hit against COG, GO, KEGG, KOG, Pfam, Swiss-Prot, TrEMBL and nr databases, respectively (Table 2). Among these unigenes, 46,413, 19,390, 35,892, 81,428, 87,457, 61,003, 109,789 and 138,584 unigenes could be annotated in the COG, GO, KEGG, KOG, Pfam, Swiss-Prot, TrEMBL and Nr databases, respectively (Table S6).
The assembled unigenes were searched against KOG database to obtain the classification of orthologous proteins. 81,428 unigenes were assigned into 25 categories (Fig. S2). The top three of which were general function prediction only (7400 unigenes), posttranslational modification, protein turnover, chaperones (4558 unigenes), and translation, ribosomal structure and biogenesis (2,904 unigenes). GO assignment was conducted to classify functions of the predicted genes of R. tanguticum (Fig. S3). Based on sequence homology, 19,390 annotated unigenes were classified into three major GO categories: biological process, cellular component and molecular function (Fig. S3).
KEGG pathway analyses can help to identify the biological pathways that are related to unigenes. In total, 35,892 annotated genes were assigned to 129 KEGG pathways. Of these pathways, top ten KEGG pathways were metabolic pathways (ko01100, 8,321 unigenes), biosynthesis of secondary metabolites (ko01110, 4710 unigenes), carbon metabolism (ko01200, 1939 unigenes), ribosome (ko03010, 1896 unigenes), protein processing in endoplasmic reticulum (ko04141, 1622 unigenes), biosynthesis of amino acids (ko01230, 1475 unigenes), spliceosome (ko03040, 1274 unigenes), RNA transport (ko03013, 959 unigenes), purine metabolism (ko00230, 901 unigenes) and oxidative phosphorylation (ko00190, 837 unigenes) (Fig. 3).

The representative KEGG pathways in the transcriptome of R. tanguticum
Identification and functional analysis of differentially expressed genes
In order to identify DEGs among the different tissues of R. tanguticum, FPKM values for each sample was calculated and normalized. In all three comparison groups (Rt_R vs. Rt_L, Rt _R vs. Rt_S and Rt_S vs. Rt_L), 196 DEGs were identified in common (Fig. 4A). Totally, the number of DEGs between the three different tissues were as follows: 2,898 between Rt_R and Rt_L (53.79% up- and 46.21% down-regulated), 2756 between Rt_R and Rt_S (58.85% up- and 41.15% down-regulated), 3147 between Rt_S and Rt_L (41.37% up- and 58.63% down-regulated) (Fig. 4B). All 5801 DEGs were recovered for hierarchical clustering analysis of transcript abundance in all three of the different tissues. The heatmap of DEGs showed the similar gene expression profiles for Rt_R, Rt_S and Rt_L (Fig. 4C).

A Venn diagram of DEGs in different comparisons. All DEGs are clustered into three comparison groups represented by three circles. The overlapping parts of different circles represent the number of DEGs in common from those comparison groups. B Number of the DEGs in different comparisons. Red represents up-regulated genes and light blue represents down-regulated genes. C Heatmap of DEGs and FPKM distribution of all genes. Each column in the figure represents one sample, each row represents one gene. The color indicates the gene expression level in the samples (log10 (FPKM + 1))
To obtain integrated functions of DEGs, all DEGs were searched against to the public databases. The results indicated that 2719 DEGs for Rt_R versus Rt_L, 2599 DEGs for Rt _R versus Rt_S and 2950 DEGs for Rt_S versus Rt_L could be annotated to COG, KOG, Pfam, Swiss-Prot, TrEMBL, and Nr database, respectively (Table S7). In addition, GO and KEGG enrichment analysis was conducted to better understand the functions of DEGs. GO significant enrichment results for different tissues were shown in Fig. S4. For the biological process category, photosynthesis (GO:0015979) (p < 0.05), plant-type cell wall organization (GO:0009664) and protein phosphorylation (GO:0006468) were top enriched terms in Rt_R versus Rt_L, Rt_R versus Rt_S and Rt_S versus Rt_L, respectively. In the cellular component category, chloroplast thylakoid membrane (GO:0009535) was top enriched in three comparative groups. For the molecular function category, we found that protein serine/threonine phosphatase activity (GO:0004674), 2-alkenal reductase [NAD(P)] activity (GO:0032440) and protein serine/threonine kinase activity (GO:0004722) were three top enriched terms in Rt_R versus Rt_L and Rt_R versus Rt_S and Rt_S versus Rt_L, respectively. We found that KEGG enrichment results were similar in different comparison groups (Fig. S5). Especially, two KEGG pathways such as starch and sucrose metabolism and plant hormone signal transduction were significantly enriched in the three comparison groups.
Genes involved in the anthraquinones biosynthesis
In this study, a total of 227 structural enzyme genes involved in the MVA, MEP, shikimate and polyketide pathways were identified, and these genes may regulate the biosynthesis of anthraquinones (Table 3). Of these genes, 100 genes encoded the enzymes involved in MVA pathway; 59 genes were related to the shikimate pathway; 18 genes were involved in the MEP pathway; 50 genes were found that encoded enzymes in polyketide pathway. Especially, we found that 8 candidate genes (PKS III-L1-8) may encode type III polyketide synthases. The phylogenetic results indicated that only one of them was clustered in the CHS group, and other genes were found in the non-CHS group (Fig. 5). As Cyt P450s (CYPs) and UDP-glycosyltransferases (UGTs) may be involved in some post-strictosamide steps of biosynthetic pathway of anthraquinones. We also screened such genes in the transcriptome of R. tanguticum. We found that 373 genes were predicted to be the members of CYP family, and 121 genes may encode UDP-Glucosyl transferase (Table 3).

Amino acid sequence-based phylogeny of the candidate enzymes in Polygonaceae belonging to type III plant polyketide synthases superfamily inferred by maximum likelihood analysis. Numbers above branches indicate the bootstrap values of each clade in the tree. The bacterial type III PKS Mycobacterium tuberculosis PKS18 was used as an outgroup
Of these candidate enzyme genes involved in the biosynthesis of anthraquinones, 14 genes (IPPS1, IPPS2, MK, SMK, EPSPs, MenE, IS, HDR, PKS III-L1, PKS III -L3, PKS III -L4, PKC1, PKC2, PKC3) were significantly differentially expressed between comparison groups. In Rt_R versus Rt_S, four genes (PKS III-L1, PKS III-L4, PKC1 and MK) showed higher gene expression levels in roots than that in stems. While the remaining five genes (SMK, MenE, IS, HDR, PKC2) were highly expressed in stems compared to that in roots (Fig. 6A). Unexpectedly, only five genes (SMK, HDR, PKC1, PKC3, PKS III -L3) were differentially expressed in Rt_R versus Rt_L, of which all had higher expression levels in leaves than that in roots (Fig. 6B). For Rt_S versus Rt_L group, eight genes (IPPS1, IPPS2, PKS III -L1, PKS III-L4, PKC1, PKC3, MK, and HDR) were highly expressed in leaves, and the remaining three genes (IS, EPSPS, PKC2) showed higher expression levels in stems than that in leaves (Fig. 6C). To validate the reliability of transcriptome sequencing results, abovementioned 14 genes involved in the anthraquinones biosynthesis were selected for qRT-PCR. The results indicated that the expression profiles of 14 genes were generally consistent with RNA-seq results (Fig. S6).

The expression level of genes involved in the biosynthesis of anthraquinones. Color changing from red to green indicates that log10 (FPKM + 1) gradually changes from high to low. A Anthraquinones-related DEGs between Rt_R and Rt_S; B Anthraquinones-related DEGs between Rt_R and Rt_L; C Anthraquinones-related DEGs between Rt_S and Rt_L
Detection of SSR loci and designation of primers
A total of 137,400 SSR loci were identified based on the transcriptome of R. tanguticum, and 28,953 unigene sequences harbored more than one SSR loci. The most common repeat types for the identified SSRs were hexanucleotides followed by trinucleotides, tetranucleotides and dinucleotides, and pentanucleotides were the least repeats in R. tanguticum transcriptome (Fig. 7). The number of repeat motifs of each locus ranged from 2 to 15, and SSRs with two repeats were the most abundant, followed by loci with four, three and five repeats. Only one SSR with 15 repeats was identified, and no SSRs with 13 or 14 repeats were available. The most abundant hexa-nucleotide repeat motif was AAAAAG/CTTTTT, followed by AAAAAT/ATTTTT and AAAAAC/GTTTTT; the number of AGCGCT/AGCGCT repeat motif was the least abundant. For the tri-nucleotide repeat units, the dominant motif was AAG/CTT, followed by AGG/CCT and AAC/GTT (Fig. 7). Based on the identified SSR loci, a total of 64,081 SSR primer pairs were successfully designed (Table S8). A total of 38 SSR primer pairs were randomly selected for the validation using agarose gel electrophoresis. The results indicated that 20 primers (52.6%) were successful in PCR amplification with genomic DNA from different Rheum species, and 13 primers showed polymorphism among these species (Fig. S7).

Frequency distribution of the SSRs identified in R. tanguticum transcriptome. A Frequency distribution of SSRs; B Total numbers of different SSR motifs. Results of hexa-nucleotides and tri-nucleotides are represented
Discussion
Comprehensive transcriptome datasets were retrieved for R. tanguticum based on the Illumina X Ten platform in current study. Illumina sequencing is superior than the Roche 454, SOLiD, and HeliScope sequencing technology, and the accuracy of Illumina sequencing have been greatly changed after the updates of technologies (Hong and Gresham 2017). Therefore, it had been widely used to analyze the structure and expression levels of transcripts and find rare or novel transcripts. Here, about 272 million of the high-quality reads were generated, and 257,942 genes were assembled based on the nine cDNA libraries of different tissues. The genes obtained from this study were much more than that from seedling transcriptomes of other Rheum species (Hei et al. 2019; Li et al. 2018). This was mainly because transcriptome datasets derived from the three tissues may provide more genetic information, and the unmatured tissues contain finite transcriptomic information due to temporality and spatiality of transcriptome. Based on the sequence annotation results, more than half of the unigene sequences (57.5%) showed significant similarity to the genes from the nr database while the remaining unigenes could not be annotated. It was because limited genomic or transcriptomic resources were available for Rheum species and even for Polygonaceae. The unigenes without annotation information may represent novel transcripts or they were matched to unknown proteins. Besides, our results showed that 86.7% of unigenes over 1000 bp in length had at least one blast hit against in the eight public databases. It has been proven that longer unigene sequences were more likely to have BLAST matches in the protein databases (Wang et al. 2012; Zhang et al. 2014; Zhou et al. 2016a, b). All in all, we are confident that the RNA-seq dataset derived from this study will be broadly useful for future studies of medicinal transcriptome dynamics in Rheum.
Anthraquinones are aromatic polyketides that could be synthesized by bacteria, fungi, insects, and plants and have various medicinal benefits (Kang et al. 2020). The detailed biosynthetic steps of anthraquinones still remains unclear for plant species. But several researches showed that a polyketide pathway and a combination of shikimate and mevalonate/methyl-D-erythritol 4-phosphate pathways were closely related to the anthraquinones biosynthesis (Kang et al. 2020; Leistner 1985; Leistner and Zenk 1968; Yamazaki et al. 2013). Based on the functional annotations, we identified large number of transcripts involved in metabolism process, biosynthesis of secondary metabolites, catalytic activity, and cellular processes. Of these transcripts, we found core genes may encode the enzymes that catalyze the biosynthetic steps in the MEP, MVA, shikimate and polyketide pathways (Table 3). In addition, more than one transcript could be assigned to the same enzyme. Plenty of genes involved in the biosynthesis of active ingredients for medicinal plants have been successfully identified by the transcriptome datasets (Vaidya et al. 2013; Wenping et al. 2011; Yuan et al. 2020). Our results further indicated that it is effective to explore the key enzyme genes related to the biosynthesis of secondary metabolites using the transcriptome sequencing. These genes will provide candidates for the genetic manipulation of anthraquinone biosynthesis in R. tanguticum. Functional classification of the candidate enzyme genes will not only help to elucidate the molecular mechanism for life saving compounds biosynthesis, but also provide cues for improving the content of these active ingredients in the future based on genetic and biochemical engineering.
CYPs were important metabolic enzymes that are involved in the biosynthesis of plant secondary metabolites. UGTs were necessary for the biosynthesis of anthraquinones, and they could catalyze glycosylation at the site of hydroxyl group and then produce glycosylated metabolites. Based on the functional annotations, 373 and 121 transcripts were predicted as putative CYPs and UGTs. However, only 125/166 CYPs and 73/66 UGTs were detected in the seedling transcriptomes of R. palmatum and R. officinale, respectively (Hei et al. 2019; Li et al. 2018). The number of these genes were less than those identified in this study. These results further indicated the ability of comprehensive transcriptomes for identifying candidate genes of secondary metabolic pathways, and these genes could be utilized for the genetic improvement of rhubarb.
Anthraquinones and their derivatives are the most effective ingredients for rhubarb. Here, we determined the content of five free anthraquinones in different tissues of R. tanguticum. Results showed that the contents of free anthraquinones in roots was higher than that in leaves and stems. Meanwhile, in the pathways that related to anthraquinone biosynthesis, the expression levels of the four genes including PKS III-L1 (Gzl_48087), PKS III-L4 (Gzl_58082), PKC1 (Gzl_37323) and MK (Gzl_70554) were detected significantly up-regulated in roots. Recently, some research speculated that a polyketide pathway is the main pathway for the biosynthesis of anthraquinones in plant species (Abdel-Rahman et al. 2013; Kang et al. 2020; Karppinen et al. 2008). In this study, eight genes might encode type III polyketide synthases were identified. Based on the phylogenetic results, only one gene PKS III-L5 (Gzl_62988) was clustered to the CHS group, and the remaining seven genes were found in the non-CHS group. We speculated that PKS III-L5 should be CHS gene in R. tanguticum, and other PKS III-Ls all belong to CHS like genes. CHS is one of type III polyketide synthases and plays important role in the biosynthesis of plant secondary metabolites such as flavonoids, stilbenes, and aromatic polyphenols (Abdel-Rahman et al. 2013; Kang et al. 2020; Pandith et al. 2016; Vadivel et al. 2018). Until now, no direct evidence was established that plant CHS enzymes catalyzed the anthraquinone biosynthesis, while several studies speculated that CHS like genes were involved in the synthesis of anthraquinones (Abdel-Rahman et al. 2013; Andersen-Ranberg et al. 2017; Kang et al. 2020; Karppinen et al. 2008). We found that PKS III-L5 was not significantly differentially expressed between roots and other two tissues in R. tanguticum. Therefore, we inferred that CHS (PKS III-L5) may also not regulate the biosynthesis of anthraquinones in R. tanguticum. However, two CHS like genes, including PKS III-L1and PKS III-L4 showed higher gene expression levels in roots than that in stems. Therefore, these two CHS like genes may contribute for the accumulation of anthraquinones in the roots. In addition, we found that most contents of free anthraquinones in leaves were higher than that in stems, and PKS III-L1 (Gzl_48087) and PKS III-L4 (Gzl_58082) were also detected significantly up-regulated in leaves, which further confirmed that CHS-like genes may also regulate the biosynthesis of anthraquinones in the leaves of R. tanguticum. PKC (Polyketide cyclase/dehydratase) is necessary for the cyclization of polyketides, which may play important roles in the formation of anthraquinones (Rama Reddy et al. 2015). Here, we found that PKC1 was highly expressed in roots and leaves, indicating PKC may also regulate the biosynthesis of anthraquinones in R. tanguticum. Unexpectedly, except for MK, most key enzyme genes in the MVA, MEP and shikimate pathway showed low gene expression levels in root. MEP pathway has been proven to occur in the plastid, so most genes related to this pathway are highly expressed in leaves (Phillips et al. 2008). Based on the expression profiles of candidate enzyme genes related to the biosynthesis of anthraquinones, we speculated that the biosynthesis of anthraquinones in roots of R. tanguticum should be primarily via polyketide pathway and regulated by CHS-like (PKS III) genes, and the anthraquinones biosynthesis in leaves and stems may be regulated by structural genes involved in MVA, MEP, shikimate and polyketide pathway. However, this conjecture is still needed to be confirmed by the enzymatic experiments in future.
Simple sequence repeat (SSR) is an important genetic marker that is commonly used for genetic breeding, genetic diversity and evolutionary studies on account of codominant and high polymorphic nature (Ali et al. 2008; Barkley et al. 2006; Van Inghelandt et al. 2010; Zhou et al. 2016a, b). Several studies have proven that transcriptome sequencing was a powerful tool for the development of SSR markers, and a large amount of SSRs developed from RNA-seq have been extensively used in plant genetic diversity analyses (Li et al. 2020; Wei et al. 2011; Xing et al. 2017; Zhao et al. 2015; Zhou et al. 2016a, b). Up to now, however, no EST-SSR resources and specific SSR primers were available for R. tanguticum. Here, a large number of SSR loci were detected based on the comprehensive transcriptome of R. tanguticum. Mononucleotide repeats were excluded for detecting SSRs since they may be resulted from base mismatch or sequencing errors, and they were difficult to distinguish from polyadenylation products (Zhou et al. 2016a, b). In addition, amount of SSR primer pairs were designed based on the identified loci, and some selected loci showed polymorphisms in different Rheum species after validation. The SSR loci in the functional regions developed in this study will not only help to enhance the genetic breeding of R. tanguticum, but also provide abundant genetic marker resources for the future genetic diversity studies for genus Rheum.
Conclusions
In this study, root, leaf and stem transcriptomes of R. tanguticum were sequenced based on Illumina sequencing platform. The resultant large numbers of unigenes provided a robust genetic basis for identifying key genes and secondary metabolic pathways for Rheum species. Base on the transcriptomic dataset, candidate enzyme genes involved in the biosynthesis of anthraquinones were identified. These genes may provide useful genetic information for the improvement of anthraquinone content in rhubarb. We also found that the high expression levels of CHS-like (PKS III) genes may be the primary cause for the content differences of anthraquinones in root and other tissues of R. tanguticum. On this basis, genetic engineering and breeding for rhubarb can be easily performed. In addition, large number of SSRs will provide abundant genetic marker resources for the future genetic diversity and evolutionary studies for Rheum species.
Availability of data and material
All the raw reads generated in this study have been deposited in the NCBI with the BioProject Accession Number of PRJNA719574.
References
Abdel-Rahman IA, Beuerle T, Ernst L, Abdel-Baky AM, Desoky EE-DK, Ahmed AS, Beerhues L (2013) In vitro formation of the anthranoid scaffold by cell-free extracts from yeast-extract-treated Cassia bicapsularis cell cultures. Phytochemistry 88:15–24
Aichner D, Ganzera M (2015) Analysis of anthraquinones in rhubarb (Rheum palmatum and Rheum officinale) by supercritical fluid chromatography. Talanta 144:1239–1244
Ali M, Rajewski J, Baenziger P, Gill K, Eskridge K, Dweikat I (2008) Assessment of genetic diversity and relationship among a collection of US sweet sorghum germplasm by SSR markers. Mol Breed 21:497–509
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Amini H, Naghavi MR, Shen T, Wang Y, Nasiri J, Khan IA, Fiehn O, Zerbe P, Maloof JN (2019) Tissue-specific transcriptome analysis reveals candidate genes for terpenoid and phenylpropanoid metabolism in the medicinal plant Ferula assafoetida. Genes Genom Genet 9:807–816
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106
Andersen-Ranberg J, Kongstad KT, Nafisi M, Staerk D, Okkels FT, Mortensen UH, Lindberg Møller B, Frandsen RJN, Kannangara R (2017) Synthesis of C-glucosylated octaketide anthraquinones in nicotiana benthamiana by using a multispecies-based biosynthetic pathway. ChemBioChem 18:1893–1897
Barkley NA, Roose ML, Krueger RR, Federici CT (2006) Assessing genetic diversity and population structure in a Citrus germplasm collection utilizing simple sequence repeat markers (SSRs). Theor Appl Genet 112:1519–1531
Chinese Pharmacopoeia Committee (2015) Pharmacopoeia of the People’s Republic of China. China Medical Science Press, Beijing
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Danielsen K, Aksnes DW, Francis GW (1992) NMR study of some anthraquinones from rhubarb. Magn Reson Chem 30:359–360
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Han Y-S, Van der Heijden R, Verpoorte R (2001) Biosynthesis of anthraquinones in cell cultures of the Rubiaceae. Plant Cell Tiss Org 67:201–220
Hei X, Li H, Li Y, Wang G, Xu J, Liang P, Deng C, Yan Y, Guo S, Zhang G (2019) High-throughput transcriptomic sequencing of Rheum officinale Baill. seedlings and screening of genes in anthraquinone biosynthesis. Chin Pharm J 54:526–535
Hong J, Gresham D (2017) Incorporation of unique molecular identifiers in TruSeq adapters improves the accuracy of quantitative sequencing. Biotechniques 63:221–226
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587
Kang S-H, Pandey RP, Lee C-M, Sim J-S, Jeong J-T, Choi B-S, Jung M, Ginzburg D, Zhao K, Won SY, Oh T-J, Yu Y, Kim N-H, Lee OR, Lee T-H, Bashyal P, Kim T-S, Lee W-H, Hawkins C, Kim C-K, Kim JS, Ahn BO, Rhee SY, Sohng JK (2020) Genome-enabled discovery of anthraquinone biosynthesis in Senna tora. Nat Commun 11:5875
Karppinen K, Hokkanen J, Mattila S, Neubauer P, Hohtola A (2008) Octaketide-producing type III polyketide synthase from Hypericum perforatum is expressed in dark glands accumulating hypericins. FEBS J 275:4329–4342
Koyama J, Morita I, Kobayashi N (2007) Simultaneous determination of anthraquinones in rhubarb by high-performance liquid chromatography and capillary electrophoresis. J Chromatogr A 1145:183–189
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Leistner E (1985) Biosynthesis of chorismate-derived quinones in plant cell cultures. Springer, Berlin, pp 215–224
Leistner E, Zenk M (1968) Mevalonic acid a precursor of the substituted benzenoid ring of Rubiaceae-anthraquinones. Tetrahedron Lett 9:1395–1396
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12:323–323
Li L, Liu K, Wei S-L, Cheng X-L, Liu J, Ren G-X, Wang W-Q (2014) Resource situation investigation about Rheum tanguticum and its sustainable utilization analysis in main production area of China. China J Chin Mater Med 39:1407–1412
Li H, Zhang N, Li Y, Hei X, Li Y, Deng C, Yan Y, Liu M, Zhang G (2018) High-throughput transcriptomic sequencing of Rheum palmatum L. seedlings and elucidation of genes in anthraquinone biosynthesis. Acta Pharm Sin 53:1908–1917
Li H, Hei X, Li Y, Liu Z, Zhang H, Gao J, Yan Y, Zhang G (2019) Accumulation profile of nine constituents in different years old stage and parts of Rheum palmatum L. by HPLC analysis. Nat Prod Res Dev 31:923–931
Li S, Ji F, Hou F, Cui H, Shi Q, Xing G, Weng Y, Kang X (2020) Characterization of Hemerocallis citrina transcriptome and development of EST-SSR markers for evaluation of genetic diversity and population structure of Hemerocallis Collection. Front Plant Sci 11:686
Liu J, Leng L, Liu Y, Gao H, Yang W, Chen S, Liu A (2020) Identification and quantification of target metabolites combined with transcriptome of two Rheum species focused on anthraquinone and flavonoids biosynthesis. Sci Rep 10:20241
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534
Mizuuchi Y, Shi SP, Wanibuchi K, Kojima A, Morita H, Noguchi H, Abe I (2009) Novel type III polyketide synthases from Aloe arborescens. FEBS J 276:2391–2401
Pandith SA, Dhar N, Rana S, Bhat WW, Kushwaha M, Gupta AP, Shah MA, Vishwakarma R, Lattoo SK (2016) Functional Promiscuity of Two Divergent Paralogs of Type III Plant Polyketide Synthases. Plant Physiol 171:2599–2619
Phillips MA, León P, Boronat A, Rodríguez-Concepción M (2008) The plastidial MEP pathway: unified nomenclature and resources. Trends Plant Sci 13:619–623
Pillai PP, Nair AR (2014) Hypericin biosynthesis in Hypericum hookerianum Wight and Arn: investigation on biochemical pathways using metabolite inhibitors and suppression subtractive hybridization. CR Biol 337:571–580
Rama Reddy NR, Mehta RH, Soni PH, Makasana J, Gajbhiye NA, Ponnuchamy M, Kumar J (2015) Next generation sequencing and transcriptome analysis predicts biosynthetic pathway of sennosides from Senna (Cassia angustifolia Vahl.), a non-model plant with potent laxative properties. PLoS ONE 10:e0129422
Ren G, Li L, Hu H, Li Y, Liu C, Wei S (2016) Influence of the environmental factors on the accumulation of the bioactive ingredients in Chinese rhubarb products. PLoS ONE 11:e0154649
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277
Rozen S, Skaletsky H (1999) Primer3 on the WWW for general users and for biologist programmers. In: Misener S, Krawetz SA (eds) Bioinformatics methods and protocols. Springer, New York, pp 365–386
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108
Shen C, Guo H, Chen H, Shi Y, Meng Y, Lu J, Feng S, Wang H (2017) Identification and analysis of genes associated with the synthesis of bioactive constituents in Dendrobium officinale using RNA-Seq. Sci Rep 7:187
Thiel T, Michalek W, Varshney R, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Vadivel AKA, Krysiak K, Tian G, Dhaubhadel S (2018) Genome-wide identification and localization of chalcone synthase family in soybean (Glycine max [L] Merr). BMC Plant Biol 18:325
Vaidya K, Ghosh A, Kumar V, Chaudhary S, Srivastava N, Katudia K, Tiwari T, Chikara SK (2013) De novo transcriptome sequencing in Trigonella foenum-graecum L. to identify genes involved in the biosynthesis of diosgenin. Plant Genome 6:1–11
Van Inghelandt D, Melchinger AE, Lebreton C, Stich B (2010) Population structure and genetic diversity in a commercial maize breeding program assessed with SSR and SNP markers. Theor Appl Genet 120:1289–1299
Wang S, Wang X, He Q, Liu X, Xu W, Li L, Gao J, Wang F (2012) Transcriptome analysis of the roots at early and late seedling stages using Illumina paired-end sequencing and development of EST-SSR markers in radish. Plant Cell Rep 31:1437–1447
Wang X, Feng L, Zhou T, Ruhsam M, Huang L, Hou X, Sun X, Fan K, Huang M, Zhou Y, Song J (2018) Genetic and chemical differentiation characterizes top-geoherb and non-top-geoherb areas in the TCM herb rhubarb. Sci Rep 8:9424
Wei W, Qi X, Wang L, Zhang Y, Hua W, Li D, Lv H, Zhang X (2011) Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom 12:451
Wenping H, Yuan Z, Jie S, Lijun Z, Zhezhi W (2011) De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 98:272–279
Xing W, Liao J, Cai M, Xia Q, Liu Y, Zeng W, Jin X (2017) De novo assembly of transcriptome from Rhododendron latoucheae Franch. using Illumina sequencing and development of new EST-SSR markers for genetic diversity analysis in Rhododendron. Tree Genet Genomes 13:53
Xu L, Dong Z, Fang L, Luo Y, Wei Z, Guo H, Zhang G, Gu YQ, Coleman-Derr D, Xia Q, Wang Y (2019) OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res 47:W52–W58
Yamazaki M, Mochida K, Asano T, Nakabayashi R, Chiba M, Udomson N, Yamazaki Y, Goodenowe DB, Sankawa U, Yoshida T, Toyoda A, Totoki Y, Sakaki Y, Góngora-Castillo E, Buell CR, Sakurai T, Saito K (2013) Coupling deep transcriptome analysis with untargeted metabolic profiling in ophiorrhiza pumila to further the understanding of the biosynthesis of the anti-cancer alkaloid camptothecin and anthraquinones. Plant Cell Physiol 54:686–696
Yu G, Wang L-G, Han Y, He Q-Y (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287
Yuan Y, Zhang J, Liu X, Meng M, Wang J, Lin J (2020) Tissue-specific transcriptome for Dendrobium officinale reveals genes involved in flavonoid biosynthesis. Genomics 112:1781–1794
Zhang W, Tian D, Huang X, Xu Y, Mo H, Liu Y, Meng J, Zhang D (2014) Characterization of flower-bud transcriptome and development of genic SSR Markers in Asian Lotus (Nelumbo nucifera Gaertn.). PLoS ONE 9:e112223
Zhao Y-M, Zhou T, Li Z-H, Zhao G-F (2015) Characterization of global transcriptome using illumina paired-end sequencing and development of EST-SSR markers in two species of Gynostemma (Cucurbitaceae). Molecules 20:21214–21231
Zhou D, Gao S, Wang H, Lei T, Shen J, Gao J, Chen S, Yin J, Liu J (2016a) De novo sequencing transcriptome of endemic Gentiana straminea (Gentianaceae) to identify genes involved in the biosynthesis of active ingredients. Gene 575:160–170
Zhou T, Li Z, Bai G, Feng L, Chen C, Wei Y, Chang Y, Zhao G (2016b) Transcriptome sequencing and development of genic SSR markers of an endangered Chinese endemic genus Dipteronia Oliver (Aceraceae). Molecules 21:166
Zhou T, Zhu H, Wang J, Xu Y, Xu F, Wang X (2020) Complete chloroplast genome sequence determination of Rheum species and comparative chloroplast genomics for the members of Rumiceae. Plant Cell Rep 39:811–824
Zhu H, Hou X, Zhang M, Zhou T, Feng L, Wang X (2021) Content determination of anthraquinone and quality evaluation of the population of source plants of rhubarb based on HPLC. Chin Tradit Herb Drugs 52:5295–5302
Acknowledgements
This study was co-supported by the National Natural Science Foundation of China (Nos. 81903739 and 31770364), the Natural Science Foundation of Shaanxi Province (Nos. 2020JQ-024 and 2020JZ-05) and the China Postdoctoral Science Foundation (No. 2018M643680).
Funding
This study was co-supported by the National Natural Science Foundation of China (Nos. 81903739 and 31770364), the Natural Science Foundation of Shaanxi Province (Nos. 2020JQ-024 and 2020JZ-05) and the China Postdoctoral Science Foundation (No. 2018M643680).
Author information
Authors and Affiliations
Contributions
TZ and XW conceived and designed the experiments. MZ collected the samples. TZ, TYZ, JS and HZ performed the experiments and analyzed the data. TZ and XW wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to participate
All authors have agreed to participate in the manuscript.
Consent to publication
All authors have agreed to publish the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Figure S1.
Length distribution of assembled unigenes (TIF 546 kb)
Supplementary Figure S2.
Classification of non-redundant unigene sequences from R. tanguticum transcriptome (TIF 1133 kb)
Supplementary Figure S3.
Gene ontology classification of non-redundant unigene sequences from R. tanguticum transcriptome (TIF 4713 kb)
Supplementary Figure S4.
GO enrichment of DEGs between comparison groups (TIF 2024 kb)
Supplementary Figure S5.
KEGG pathway enrichment of DEGs between comparison groups (TIF 697 kb)
Supplementary Figure S6.
Verification and expression pattern analysis on 14 genes involved in pathway for anthraquinones biosynthesis. A Expression levels of 14 genes from RNA-seq (log2(FPKM + 1)); B Relative expression levels of 14 genes from qRT-PCR (TIF 916 kb)
Supplementary Figure S7.
Validation of a subset of the SSR primer pairs for the Rheum species by agarose-gel profiling. Results are shown for one individual from each species with primer name on top; Abbreviated species names were shown on schematic diagrams: Ro, Rheum officinale; Rt, R. tanguticum, Rtt, R. tanguticum var. liupanshanense; Rp, R. palmatum; M, DL2000 DNA ladder (TIF 4464 kb)
Supplementary Table S1.
Candidate type III polyketide synthases recovered from GenBank (XLSX 16 kb)
Supplementary Table S2.
Primers for the qRT-PCR validation (XLSX 10 kb)
Supplementary Table S3.
Thirty-eight SSR primer pairs use for PCR validation (XLSX 13 kb)
Supplementary Table S4.
The content differences in five free anthraquinones between the three different tissues of R. tanguticum showed by ANOVA one-way analysis of variance (XLSX 16 kb)
Supplementary Table S5.
Reads information of nine cDNA libraries from three different tissues (DOCX 15 kb)
Supplementary Table S6.
Integrated annotation information of all non-redundant unigenes in eight databases (XLSX 15750 kb)
Supplementary Table S7.
Integrated annotation information of DEGs between three different tissues of R. tanguticum (XLSX 881 kb)
Supplementary Table S8.
SSR primer pairs designed based on the candidate SSR loci in transcriptome of R. tanguticum (XLSX 5969 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Zhou, T., Zhang, T., Sun, J. et al. Tissue-specific transcriptome for Rheum tanguticum reveals candidate genes related to the anthraquinones biosynthesis. Physiol Mol Biol Plants 27, 2487–2501 (2021). https://doi.org/10.1007/s12298-021-01099-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-021-01099-8