
Abstract
Horsegram is an important drought resistant pulse crop from Fabaceae and can be easily grown in dry lands with no irrigation facilities. However, it remained neglected since long and has been considered as orphan legume which requires immediate attention for its improvement and for the development of new promising varieties in future. In the present study, 7352 simple sequence repeat (SSR) markers were developed from the transcriptome data and 150 SSR were randomly synthesized for validation and diversity analysis in a panel of 58 horsegram genotypes. The synthesized primers included all types of repeats spanning direpeats to hexarepeats. Of the validated SSR markers, 33 markers were polymorphic and produced 40 loci which were used to analyze the genetic diversity and structure of horsegram. In total, 130 alleles were produced in a range of 2–9 alleles with maximum alleles produced by primer HTSSR 155. Expected heterozygosity (He) ranged from 0.03 to 1.00 and observed heterozygosity (Ho) ranged from 0.13 to 0.81. Polymorphism information content value ranged from 0.065 to 0.78. Dendrogram based on UPGMA and principal component analysis showed four groups of the 58 genotypes of horsegram. Structure analysis showed three genetic stocks for the analyzed germplasm. Thus, the developed SSRs can be useful in future population genetics analysis, molecular breeding studies and mapping works in horsegram germplasm as well as in related legume species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Horsegram [Macrotyloma uniflorum Lam. (Verdc.)] is a protein rich, underutilized, ancient Indian pulse crop of family Fabaceae (Prasad and Singh 2015; Fuller and Murphy 2018). It is commonly known as kulath, kulthi, ulavalu, hurali, kollu or muthira etc. in various parts of India. Its English name was derived from its feeding to horses (Bhardwaj et al. 2013). It is a rich source of proteins, minerals and vitamins. Besides its nutritional importance, it has been found to possess medicinal properties, because of the presence of some non-nutritive bioactive substances. It is considered as a pulse with medical values (Bolbhat and Dhumal 2014; Bhartiya et al. 2015; Vandarkuzhali and Narayanasamy 2015; Fuller and Murphy 2018). It possesses anti-hypercholesterolemic, anti-microbial, anti-obesity, anti-helminthic, analgesic, anti-inflammatory, anti-diabetic, anti-cholilithiatic, anti-histaminic, anti-peptic ulcer, anti-oxidant, anti-urolithiatic, diuretic, haemolitic, hepatoprotective, and anti-hypertensive properties (Ranasinghe and Ediriveera 2017).
In the developing countries like India, only a few conventional legumes dominate the pulse production. Therefore, to effectively eradicate the protein malnutrition, the underutilized legumes like horsegram have great potential for improving nutritional security of rural, tribal and underprivileged people (Tontisirin 2014). The crop accounts for about 5–10% of pulse production in India and its annual production is about 0.65 million tonnes (Kiranmai et al. 2016). Horsegram has also been identified as a potential food source for the future. It is also used as cattle feed in both fresh and dried form. It is known to be useful for water conservation in the semi-arid region because of low water requirement. Therefore, it is widely grown in the semi-arid regions of India. It is generally considered as protein rich poor man’s crop that grows well under dry conditions and marginal soil fertility (Kiranmai et al. 2016). It is now established that horsegram also performs better than many other pulse crops under saline conditions (salt stress conditions), therefore this crop has a higher production potential under such conditions (Reddy et al. 2008).
Despite its importance, the crop has been neglected for long and has not received its due recognition in research fraternity, however, recently this trend seems to be reversing. Studies on characterization of horse gram germplasm have been carried out using morphological markers. However, few studies have utilized molecular markers for the characterization of horse gram germplasm. Few mutation studies have also been carried out in the direction of understanding some characters in the crop. Since this crop comes up reasonably well in dry land areas with receding soil moisture conditions and in poor soils where other crops fail to grow, there is also high probability that this crop contain a large number of drought resistance genes. However, over the years, the production and area under this crop has been decreasing tremendously owing to non-availability of improved and well adapted varieties. The studies on morphological traits diversity have been conducted in this crop by different workers and they have suggested a great potential for its improvement (Geetha et al. 2011; Neelam et al. 2014; Singh et al. 2019).
Although few molecular studies have been carried out recently in this crop but SSRs have not been utilized to a greater extent in this important food and fodder crop (Ramya et al. 2013; Kiranmai et al. 2018). This may be due to limited availability of genetic and genomic resources in this crop. Therefore, it is immensely important to develop SSR marker resources in this crop which could be used to study genetic diversity, linkage analysis, QTL and association mapping studies as demonstrated by other workers in various crops (Qiu et al. 2010; Wang et al. 2011, 2013; Xue et al. 2018; Chahota et al. 2020). In addition, marker-assisted selection (MAS) and genomic selection (GS) can also be performed if sufficient genomic resources become available for the purpose. So far, limited numbers of SSRs have been developed and characterized in this crop. Sharma et al. (2015a, b) have identified and developed 245 SSR and 13 Intron Length Polymorphism (ILP) markers from public sequence data. Similarly, Divya (2015) and Kaldate et al. (2017) employed public sequence data to design and validate SSR markers in this crop. Chahota et al. (2017) used next-generation Illumina sequencing platform to develop a large number of microsatellite markers in this species. Of the total 23,305 potential SSRs motifs, 5755 primers were designed by their group. Of these designed primers, 30 SSRs were used in 360 accessions to study genetic diversity and population structure. However, more marker resources are required in this species to initiate genetic improvement programmes and apply genomic tools in combination with other conventional techniques which can be a promising strategy of improvement (Datir 2016). Simple sequence repeats (SSRs) are widely used due to their codominant inheritance, multi-allelic nature, high reproducibility and transferability, extensive genome coverage, and simple detection methods (Choudhary et al. 2009; Sharma et al. 2009; Kaur et al. 2016). Therefore, in present study we identified, developed and utilized SSR markers in this crop to enrich genomic resource data and to analyze genetic structure in a panel of horsegram genotypes. Furthermore, the SSRs identified and characterized in this study were from drought resistant genotypes and hence, can also be useful in identification and screening of drought resistant accessions in future.
Materials and methods
SSR markers development and gene ontology
Transcriptome database was used to develop new SSRs. A total of 110.2 MB clean, filtered, FASTQ sequences of horsegram were acquired from NCBI SRA (sequence read archive) under accession numbers SRX341972, SRX341973, SRX341974 and SRX341975. Reads were assembled into 124,147 contigs using de novo assembly module of CLC Genomics Workbench v 6.5 (CLC Inc, Aarhus, Denmark) with overlap length cut off value of 150 and N50 value of 851. Assembled contigs were utilized for transcriptomic SSR mining using MISA tool with criteria of direpeats with minimum six units and trirepeats, tetrarepeats, pentarepeats and hexarepeats having minimum five units with the maximum difference between two SSRs setting at 100 bp.
All SSR-containing sequences were utilized for primer design following standard parameters by using BatchPrimer3 software (https://probes.pw.usda.gov/cgibin/batchprimer3/batchprimer3.cgi) considering the following criteria: (1) primer length ranging from 18–22 bp with an optimum 20 bp, (2) PCR product size ranging from 100–400 bp with an optimum 300 bp, (3) annealing temperature ranging from 45–60 °C, (4) GC content ranging from 40–60 with an optimum of 50%. Newly designed transcriptomic SSRs were named as horsegram transcriptomic SSRs (HTSSR).Gene ontology classification of SSR containing sequences assigned 13,427 GO terms. Open Reading Frames (ORF) were identified in SSR containing sequences using ORF finder. Earlier SSR work (Sharma et al. 2015a, b) did not include this information in their studies. In-House pearl script based programme was used to find the location of SSRs in SSR containing sequences. Further, KEGG pathways analysis was performed on 6211 SSR containing sequences.
Plant materials and genomic DNA isolation
Based on the evaluation made for early flowering and maturation period, a panel of diverse 58 horsegram genotypes (Table 1) collected from different horsegram growing geographical regions of the country and maintained at DAV University, Jalandhar was selected based on the flowering period and maturation timing in Jalandhar region of Punjab. Genomic DNA was isolated from young leaves of each plant using the modified CTAB method (Doyle and Doyle 1990; Rana et al. 2017) with some modifications. The quantity and quality of DNA was estimated through electrophoresis using 1 per cent agarose gel by comparing with lambda DNA (Fermentas, Lithuania).
PCR amplification
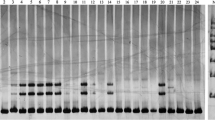
For amplification of genomic DNA, a reaction mixture of 10 µl volume was prepared using 4.8 µl of sterilized distilled water, 2.0 µl genomic DNA (13 ng/ µl), 0.5 µl of forward and 0.5 µl of reverse primer (5 µM), 0.5 µl MgCl2 (25 mM), 1.0 µl 10× PCR buffer (10 mM Tris–Hcl, 50 mM Kcl, pH 8.3), 0.5 µl dNTP mix (0.2 mM each of dATP, dGTP, dCTP and dTTP) and 0.2 µl Taq polymerase (5 U/µl).The PCR conditions were: 1 cycle of 5 min at 94 °C, 35 cycles of 1 min at 94 °C, 1 min at respective annealing temperature for each primer, 1 min at 72 °C, final extension for 7 min at 72 °C and storage at 4 °C for ∞. All the PCR reactions were conducted in 96 well Thermal Cycler Veriti™ (Applied Biosystems, CA, USA), respectively. The PCR products were first checked on 3% agarose gel and then resolved in 6% polyacrylamide gel at a constant current of 65 W at room temperature for 90 min. Gels were prepared and run in 1× TBE buffer and visualization of fragments was done using silver-staining. Size estimation of the alleles generated by newly developed markers was done by using 50 bp DNA size standard.
Data analysis
All SSR primer generated fragments were scored manually for each SSR locus; polymorphic bands were scored and converted into binary data as 1 for presence or 0 for absence of the bands respectively. Only unambiguously amplified alleles were scored and included for further analysis. The polymorphism information content (PIC) is a measure of the effectiveness of given DNA marker for detecting polymorphism. The PIC for each primer pair was calculated according to the following standard formula given by Botstein et al. (1980) and implemented in Cervus version 3.0.
where Pij is the frequency of the jth pattern for marker i and summation extends over n patterns. Various genetic diversity estimates such as expected heterozygosity (He), observed heterozygosity (Ho), Shannon information index (I), etc. were calculated with the help of POPGENE version 1.32 (Yeh and Boyle 1997). Distance-based cluster analysis was performed by generating dendrogram based on unweighted pair-group method of arithmetic mean (UPGMA) using Jaccards similarity coefficient with the help of NTSYS pc-2.02e (Rohlf 1998). Neighbour-Joining (NJ) tree was constructed using Jaccards coefficient with the help of DARwin Version 6.0.20 accessed on 28th Mrach 2019 (Perrier and Jacquemoud-Collet 2006). Genetic relationships among the genotypes were also analysed by principal component analysis (PCA). Genetic structure analysis was performed by the Bayesian clustering model using software STRUCTURE version 2.3 (Pritchard et al. 2000). An admixture model with correlated allele frequencies was used to infer the value of K with prior population information. All analyses were performed with a burn-in period of 1,00,000 and a Markov chain Monte Carlo (MCMC) replication number set at 10,00,000. The value of K was estimated using the method described by Evanno et al. (2005) and was obtained using STRUCTURE HARVESTER (Earl and VonHoldt 2012). Analysis of molecular variance (AMOVA) was done using GenAlEx (Peakall and Smouse 2012).
Results and discussion
With the advancing technologies, ease to access and exploration of plant systems has increased. Same is the case of generating SSR markers. It was very laborious and time consuming process when isolating SSR using conventional methods. However, with the advent of Next generation sequencing (NGS) techniques it becomes easy and lowcost affair to develop sequence based molecular markers like EST-SSRs. Plenty of sequences can be produced through NGS techniques in a very short time resulting in huge amount of data generation. This sequence data can be utilized to develop SSR markers as also developed by several workers in various crops (Zalapa et al. 2012; Ravishankar et al. 2017; Neophytou et al. 2018; Tibihika et al. 2019; Patil et al. 2020). We also utilized horsegram data from public domain to develop SSR markers, specifically from the drought resistant transcripts.
SSR designing and gene ontology
In toto, 7352 SSR primers were designed from the mined sequences of horsegram transcriptome. Of these, 1785 (24%) were direpeats, 4380 (59%) were trirepeats, 584 (8%) were tetrarepeats, 258 (3.5%) were pentarepeats and 345 (4.6%) were hexarepeats. Overall, frequency of SSR occurrence was observed ~ 6% (Table 2). In earlier studies, SSR frequencies were reported between 2.65 and 16.8% in 49 dicotyledonous species (Kumpatla and Mukhopadhyay 2005). Similarly, di-, tri- and tetra-nucleotide repeat containing EST-SSR frequency observed in monocot species was 1.5 and 4.7 (Kantety et al. 2002) and 7–10% (Varshney et al. 2002). The GO terms were classified into three categories namely (1) biological process 38.69% (5196) followed by (2) molecular function 34.87% (4682) and (3) cellular component 26.43% (3549). In biological category, genes involved in cellular and metabolic processes were found to be the most prevailing. In molecular function, DNA binding proteins, catalytic proteins and transcription regulators were found to be most abundant. It indicates that molecular processes involving DNA such as replication, transcription and DNA modifications may be methylation or acetylation are prominent. In addition, catalytic proteins regulating and speeding up many molecular processes also dominate other molecules. Whereas, in cellular component, majority of the genes were involved in cell, cell parts or organelle structure and function (Fig. 1). Survey of ORF showed that majority of SSRs (57%) were found in functional coding sequence region (CDS) followed by SSRs in 3' UTR (22%) and 5' UTR (16%). SSRs in inter 5'CDS regions and inter 3'CDS were found to be the least (Fig. 2). This will be helpful for many genetic manipulation works in future using new technologies to enhance the adaptability or quality of the crop. Further, KEGG pathway analysis performed on 6211 SSR containing sequences generated 1800 KEGG IDs corresponding to 152 pathways. These IDs also corresponded to 1483 enzymes in six categories as classified by enzyme commission (EC). Thus, the SSRs analyzed in present study have many implications in the different processes and pathways taking place in the plant. The details of newly designed primers are given in supplementary file 1.

Classification and functional annotation of SSR markers

Localization of SSR regions in ORFs and their vicinity and EC classification
Polymorphic extent of markers
From the 150 randomly synthesized SSR markers, 33 markers were polymorphic and produced 40 loci (Table 3). In total, 130 alleles were produced in a range of 2–9 alleles with an average of 3.25 per locus. Maximum numbers of alleles (9) were produced by primer HTSSR 155 while minimum numbers of alleles (2) were produced by nineteen different primers. Observed heterozygosity (Ho) ranged from 0.03 to 1.00 with an average of 0.55 and expected heterozygosity (He) ranged from 0.13 to 0.81 with an average of 0.56. PIC value ranged from 0.065 to 0.78 with an average of 0.47 (Table 4). The values shows that some of the primers were highly informative as compared to previous reports (Divya 2015; Sharma et al. 2015a, b; Chahota et al. 2017; Kaldate et al. 2017). These selected primers can be useful for exploring the highly conservative germplasm of horsegram in some geographical regions.
Diversity and cluster analysis
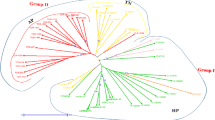
The cluster analysis of 58 accessions showed four major clusters with high (> 50.0%) bootstrap value in N-J tree based on Jaccards coefficient and principal component analysis (Figs. 3, 4). Each clustering method supported one another. Group one was further sub-classified into two sub-groups: Group 1A and Group 1B. Group 1A had genotypes procured from NBPGR, Hyderabad. In Group 1B, accessions procured from NBPGR, Uttrakhand clustered together. Based on our field evaluation and observations, Group 1A included early maturation accessions (average duration of 77 days of Monsoon season) while Group 1B included accessions exhibiting late maturation (average duration of 63 days of Monsoon season). Group 2 was found to be a small group containing 3 accessions collected from Himachal Pradesh producing early maturation (average duration of 62 days of Monsoon season). Group 3 was further sub classified into two subgroups, Group 3A and Group 3B. Group 3A included accessions procured from NBPGR Uttrakhand, giving early maturation in monsoon season (average duration of 49 days of Monsoon season). While group 3B included accessions from NBPGR Hyderabad, showing late maturation (average duration of 93 days of Monsoon season). Group 4 clustered genotypes from different geographical locations of Himachal Pradesh primarily showing late maturation (average duration of 90 days of Monsoon season). In the present study, N-J tree analysis exhibited apparent groupings based on the geographical source of horsegram accessions in addition to clear demarcation for early and late maturation of the crop in their respective categories. The analysis of N-J tree and its findings were found coherent with the Bayesian model based cluster analysis which was used to check genome sharing among studied accessions.

Principal component analysis (PCA) of 58 horsegram genotypes based on 130 fragments amplified by novel 33 SSRs

Neighbour-Joining (N-J) tree of 58 horsegram genotypes constructed using Jaccards coefficient using 130 fragments amplified by novel 33 SSRs
Bayesian model based cluster analysis
Bayesian clustering method is a powerful computational tool meant for estimating various features of populations. STRUCTURE assumes K (unknown) populations for the given data set, and the value of K can be estimated by posterior probability of the data for a given K. Delta K, which is used to determine the best fit value of K, was computed by STRUCTURE HARVESTER for the given range, i.e. 1–10 and highest value was shown at K = 3. The numbers of populations (K) were identified by performing five independent run by setting the value of K from 1–10 with a burn-in period of 1,00,000 and 10,00,000 number of Markov Chain Monte Carlo (MCMC) repeats after burn-in. Based on the maximal value of Ln P (D), Posterior probability of data using STRUCTURE HARVESTER software, a clear delineation of K was found to be 3 (Fig. 5, Supplementary Table 1). At K = 3, it was clearly possible to classify the 58 horsegram genotypes in 3 clusters irrespective of the ones with ‘pure’ ancestry or ‘mixed’ ancestry (Fig. 5). The percentage of accessions belonging to pure ancestry (accessions with membership probabilities ≥ 80%) were found to be 66, 59 and 50% in cluster one, two and three respectively indicating a strong genetic structure in the analyzed germplasm of the crop. This shows the mixing of three genetic stocks in the past was not prominent and different germplasm stocks can be utilized to produce fruitful results in future breeding programmes. At least 23 (40%) tested horsegram accessions belonged to the ‘pure’ ancestry spanning all three clusters. Rest of the accessions had shared/mixed ancestry. A comparison of the results from Bayesian-based STRUCTURE analysis with the NJ based tree revealed considerable congruence. The structure analysis showed three genetic stocks for the analyzed germplasm of horsegram which is in contradiction to earlier studies by different workers (Sharma et al. 2015a, b; Chahota et al. 2017; Kaldate et al. 2017). This may be due to diverse collection of genotypes or drought specific SSR markers used in this study. However, it can be further validated using larger numbers of genotypes countrywide in future. Result of this diversity analysis suggested that we have diverse set of horsegram germplasm which can be used in future improvement programmes and the novel polymorphic SSRs developed in this study can be useful for various genetic studies in Horsegram and related legumes.

a Posterior probability based on LnP (D) values detected by 33 novel microsatellites using STRUCTURE HARVESTER software showing a clear delineation of 3 gene pools (K = 3) in 58 accessions of Horsegram. b Bar plot showing genetic structure of 58 horse gram accessions as inferred by STRUCTURE v2.3.3
References
Bhardwaj J, Chauhan R, Swarnkar MK, Chahota RK, Singh AK, Shankar R, Yadav SK (2013) Comprehensive transcriptomic study on horsegram (Macrotyloma uniflorum): de novo assembly, functional characterization and comparative analysis in relation to drought stress. BMC Genom 14:647
Bhartiya A, Aditya JP, Kant L (2015) Nutritional and remedial potential of an underutilized food legume horsegram (Macrotyloma uniflorum): a review. J Animal Plant Sci 25(4):908–920
Bolbhat SN, Dhumal KN (2014) Mineral constituents in seeds of horsegram mutants. Int J Adv Sci Tech Res 4(3):2249–9954
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32(3):314–331
Chahota RK, Divya S, Rana M, Sharma V, Nag A, Sharma TR, Rana JC, Hirakawa H, Isobe S (2017) Development and characterization of SSR markers to Study genetic diversity and population structure of horsegram germplasm (Macrotyloma uniflorum). Plant Mol Biol Rep 35(5):550–561
Chahota RK, Sharma V, Rana M, Sharma R, Chaudhary S, Sharma TR, Shirasava K, Hirakava H, Isobe SN (2020) Construction of a framework linkage map and genetic dissection of drought- and yield-related QTLs in horsegram (Macrotyloma uniflorum). Euphytica 216:61
Choudhary S, Sethy NK, Shokeen B, Bhatia S (2009) Development of chickpea EST-SSR markers and analysis of allelic variation across related species. Theor Appl Genet 118:591–608
Datir SS (2016) Genetic improvement in horse gram [(Macrotyloma uniflorum (Lam.) verdec.] (syn. Dolichos biflorus L.) through induced mutations. J Food Legumes 29:174–179
Divya S (2015) Analysis of genetic diversity and population structure in horsegram (Macrotyloma uniflorum) germplasm using SSR markers. MSc thesis, Department of Agricultural Biotechnology, CSK Himachal Pradesh Krishi Vishvavidyalaya, Palampur, India, p 87
Doyle JJ, Doyle JE (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Earl DA, VonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Fuller DQ, Murphy C (2018) The origins and early dispersal of horsegram (Macrotyloma uniflorum), a major crop of ancient India. Genet Resour Crop Evol 65:285–305
Geetha K, Mani AK, Hepziba MJ, Latha R, Shanthi P (2011) Studies on genetic diversity among germplasm accessions of horsegram Macrotyloma uniflorum (Lam.) Verdec. Legume Res Int J 34:14–19
Kaldate R, Rana M, Sharma V, Hirakawa H, Kumar R, Singh G, Chahota RK, Isobe SN, Sharma TR (2017) Development of genome-wide SSR markers in horsegram and their utilization for genetic diversity and cross-transferability analysis. Mol Breed 37:103
Kantety RV, La Rota M, Matthews DE, Sorrells ME (2002) Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol Biol 48:501–510
Kaur K, Sharma V, Singh V, Saleem Wani MS, Gupta RC (2016) Development of novel SSR markers for evaluation of genetic diversity and population structure in Tribulus terrestris L. (Zygophyllaceae). 3 Biotech 6(2):156
Kiranmai K, Gunupuru LR, Nareshkumar A, Reddy VA, Lokesh U, Pandurangaiah M, Venkatesh B, Kirankumar TV, Sudhakar C (2016) Expression analysis of WRKY transcription factor genes in response to abiotic stresses in horsegram (Macrotyloma uniflorum (Lam.) Verdc.). Front Plant Sci 16(9):346
Kiranmai K, Rao GL, Pandurangaiah M, Nareshkumar A, Reddy VA, Lokesh U, Venkatesh B, Johnson AMA, Sudhakar C (2018) A novel WRKY transcription factor, MuWRKY3 (Macrotyloma uniflorum Lam. Verdc.) enhances drought stress tolerance in transgenic groundnut (Arachis hypogaea L.) plants. Front Plant Sci 9:346
Kumpatla SP, Mukhopadhyay S (2005) Mining and survey of simple sequence repeats in expresses sequence tags of dicotyledonous species. Genomics 48:985–998
Neelam S, Kumar V, Natarajan S, Venkateshwaran K, Pandravada SR (2014) Evaluation and diversity observed in horsegram (Macrotyloma uniflorum (Lam.) Verdc.) germplasm from Andhra Pradesh, India. Int J Plant Res 4(1):17–22
Neophytou C, Torutaeva E, Winter S, Meimberg H, Hasenauer H, Curto M (2018) Analysis of microsatellite loci in tree of heaven (Ailanthus altissima (Mill.) Swingle) using SSR-GBS. Tree Genet Genomes 14:82
Patil PG, Singh NV, Parashuram S, Bohra A, Mundewadikar DM, Sangnure VR, Babu KD (2020) Sharma J (2020) Genome wide identification, characterization and validation of novel miRNA-based SSR markers in pomegranate (Punica granatum L.). Physiol Mol Biol Plants 26:683–696
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in excel. Population genetic software for teaching and research an update. Bioinformatics 28:2537–2539
Perrier X, Jacquemoud-Collet JP (2006) DARwin software. https://darwin.cirad.fr/darwin. Accessed on 28 Mar 2019
Prasad SK, Singh MK (2015) Horse gram—an underutilized nutraceutical pulse crop: a review. J Food Sci Technol 52(5):2489–2499
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qiu LJ, Yang C, Tian B, Yang JB, Liu AH (2010) Exploiting EST databases for the development and characterization of EST-SSR markers in castor bean (Ricinus communis L.). BMC Plant Biol 10:278
Ramya M, Reddy KE, Sivakuma M, Pandurangaiah M, Nareshkumar A, Sudhakarbabu O, Veeranagamallaiah G, C, Sudhakar (2013) Molecular cloning, characterization and expression analysis of stress responsive dehydrin genes from drought tolerant horsegram (Macrotyloma uniflorum (Lam.) Verdc.). Int J Biotech Biochem 9(3):293–312
Rana JC, Rana M, Sharma V, Nag A, Chahota R, Sharma TR (2017) Genetic diversity and structure of pea (Pisum sativum L.) germplasm based on morphological and SSR markers. Plant Mol Biol Rep 35(1):118–129
Ranasinghe RLDS, ERHSS, Ediriveera (2017) Medicinal and nutritional values of Macrotyloma uniflorum (Lam.) verdc. (kulattha): a conceptual study. Glob J Pharmaceu Sci. 1(2):555–559
Ravishankar KV, Vasudeva R, Hemanth B, Sandya BS, Sthapit BR, Parthasarthy VA, Rao VR (2017) Isolation and characterization of microsatellite markers in Garcinia gummi-gutta by next-generation sequencing and cross-species amplification. J Genet 96:213–218
Reddy PCO, Sairanganayakulu G, Thippeswamy M, Reddy PS, Reddy MK, Sudhakar C (2008) Identification of stress-induced genes from the drought tolerant semi-arid legume crop horse gram [Macrotyloma uniflorum (Lam.) Verdc.] through analysis of subtracted expressed sequence tags. Plant Sci 175(3):372–384
Rohlf FJ (1998) NTSYS-pc numerical taxonomy and multivariate analysis system, version 2.0. Exeter Publishing, Ltd., Setauket
Sharma V, Bhardwaj P, Kumar R, Sharma RK, Sood A, Ahuja PS (2009) Identification and cross-species amplification of EST derived SSR markers in different bamboo species. Conserv Genet 10:721–724
Sharma V, Rana M, Katoch M, Sharma PK, Ghani M, Rana JC, Sharma TR, Chahota RK (2015) Development of SSR and ILP markers in horsegram (Macrotyloma uniflorum), their characterization, cross-transferability and relevance for mapping. Mol Breed 35:102
Sharma V, Sharma TR, Rana JC, Chahota RK (2015) Analysis of genetic diversity and population structure in horsegram (Macrotyloma uniflorum) using RAPD and ISSR markers. Agric Res 4:221–230
Singh R, Salam JL, Mandavi NC, Saxena RR, Sao A (2019) Genetic diversity estimation in horsegram [Macrotyloma uniflorum (L.) Verdcout] genotypes collected from bastar plateau. Int J Curr Microbiol App Sci 8(12):613–620
Tibihika PD, Curto M, Dornstauder-Schrammel E, Winter S, Alemayehu E, Waidbacher H, Meimberg H et al (2019) Application of microsatellite genotyping by sequencing (SSR-GBS) to measure genetic diversity of the East African Oreochromis niloticus. Conserv Genet 20:357–372
Tontisirin K (2014) Promotion of underutilized indigenous food resources for food security and nutrition in Asia and the Pacific. In: Durst P, Bayasgalanbat N (eds) Promotion of underutilized indigenous food resources for food security and nutrition in Asia and the Pacific. RAP Publications, Bangkok, pp 21–25
Vandarkuzhali P, Narayanasamy S (2015) Horse gram—a prospective functional food. Ind J Nutri Dietet 52(2):226–243
Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol Biol Lett 7:537–546
Wang Z, Yan HW, Fu XN, Li XH, Gao HW (2013) Development of simple sequence repeat markers and diversity analysis in alfalfa (Medicago sativa L.). Mol Biol Rep 40:3291–3298
Wang ZY, Li J, Luo ZX, Huang LF, Chen XL, Fang B, Li Y, Chen J, Xhang X (2011) Characterization and development of EST-derived SSR markers in cultivated sweet potato (Ipomoea batatas). BMC Plant Biol 11:139
Xue H, Zhang P, Shi T, Yang J, Wang L, Wang S, Su Y, Zhang H, Oiao Y, Li X (2018) Genome-wide characterization of simple sequence repeats in Pyrus bretschneideri and their application in an analysis of genetic diversity in pear. BMC Genomics 19:473
Yeh FC, Boyle TJB (1997) Population genetic analysis of codominant and dominant markers and quantitative traits. Belg J Bot 129:157
Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, Simon P (2012) Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot 99:193–208
Acknowledgments
Dr. Rahul Kumar acknowledges the financial grant received from DST-SERB, New Delhi in the form of young scientist research project. NBPGR, New Delhi is also duly acknowledged for providing the germplasm of Horsegram. Dr. Rahul Kumar is also thankful to Dr. Poonam Suri, Chancellor, DAV University and President, DAVCMC New Delhi for providing logistics support at DAV University, Jalandhar to conduct and complete the current research work.
Funding
This study was funded by DST-SERB, Government of India (YSS/2015/00521).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, R., Kaundal, S.P., Sharma, V. et al. Development of transcriptome-wide SSR markers for genetic diversity and structure analysis in Macrotyloma uniflorum (Lam.) Verdc.. Physiol Mol Biol Plants 26, 2255–2266 (2020). https://doi.org/10.1007/s12298-020-00898-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-020-00898-9