
Abstract
The present investigation aimed to explore the level of genetic diversity, determine the population structure in a larger set of germplasm of linseed using microsatellite marker and identify linked markers through association mapping. A total of 168 accessions of linseed were evaluated for major agro-economic traits and SSRs markers deployed for diversity assessment. A total of 337 alleles were amplified by 50 SSRs ranging from 2 to 13 with an average of 6.74 ± 2.8 alleles per loci. The neighbor joining based clustering grouped all the accessions into three major clusters that were also confirmed by scatter plot of PCoA. While model based clustering determined four sub-populations (K = 4). Further, analysis of molecular variance analysis considering three population showed that maximum variation (79%) was within the population. We identified one putative SSR marker (Lu_3043) linked with days to 50% flowering through both GLM and MLM analysis of association mapping. The results of this preliminary study revealed genetic diversity, population structure in linseed and linked marker which could be utilized in future breeding program.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Flax or Linseed (Linum usitatissimum L., 2n = 30) is an annual self-pollinated and oldest among the domesticated plants. It belongs to the family Linaceae that comprises 22 genera and 300 species (Hickey 1988). The centre of origin of flax has been proposed to be the Middle East, Mediterranean basin, Ethiopia and India (Vavilov 1926; Zohary and Hopf 2000). Flax is considered as a ‘‘founder crops’’ that has been providing raw materials for medicine, food and textiles for more than 8000 years and is of great importance to the human welfare (Zeist and Bakker-Heeres 1975). Flax seed is a good source of lignans that have anticancer properties (Westcott and Muir 2003). The high content of omega -3 fatty acid (α-linolenic acid, 55–65%) in the oil is the characteristic of linseed that make it distinct from other oil seed plants. Due to quick drying properties, it’s oil has huge industrial applications including preparation of paints, varnishes, soap, putty and polymers. Through mutation breeding approach altered fatty acid profiles linseed lines, such as low linolenic acid (2–4%) and high linoleic acid (>50%), have been developed for food purposes (Green et al. 2008). Additionally, fibre industries developed high value products from linseed for applications in automobile, construction industries, biofuel industries and pulp in last decade (Diederichsen and Ulrich 2009). It has been suggested that the cultivated flax is descended from a single domestication event from L. bienne for its oil. Molecular studies have also supports the domestication process from L. bienne, to oil and fibre flax (Allaby et al. 2005). Since, flax has divergent breeding history and a complex domestication process; it is assumed that it has limited gene flow and complex population structure.
To begin with strategic crop improvement programs, knowledge about the extent of genetic diversity and population structure of the concern crop laid an important foundation. Population structure analysis is also important for association mapping as it is essential to define the population stratification within the accessions to avoid any false association (Flint-Garcia et al. 2005). The current global prospect and health related properties of linseed have dictated the need for the evaluation of its genetic diversity through molecular markers and exploitation by marker assisted breeding (Pali et al. 2015, 2014; Chandrawati et al. 2014; Rajwade et al. 2010; Kale et al. 2012). However, informations on large genetic stocks of linseed especially that developed and grown in India is available from only a few reports of AFLP, ISSR and RAPD markers. Considering the importance of the crop and limited marker based studies till date, we have applied the SSR marker on a large set of genetic material to assess the level of diversity, determine the population structure and performed association analysis.
Materials and methods
Plant material
The plant material used in the present study includes 168 accessions of linseed obtained from All India Co-ordinated Research Program on Linseed (AICRP on Linseed), Chandra Shekhar Azad University of Agriculture and Technology (CSAUA&T), Kanpur, Uttar Pradesh, India. Of these 168 accessions, 58 are designated as germplasm lines that include prominent and popular varieties, advance breeding lines developed in India. The rest 110 accessions designated as core-germplasm include few global collections also. All the accessions were grown in field during the crop season November 2014–March 2015 at CSIR-National Botanical Research Institute, Lucknow for morphological characterization. All the entries were planted in Randomized Block Design (RBD) with 3 replications. Four rows of 2 m with spacing of 45 cm between rows and 15 cm between plants were adopted with two non experimental border rows. Normal cultural practices were followed to raise a good crop. Ten plants in each replication were randomly tagged to record data on various morphological traits including days of 50% flowering (DOF), plant height (PH), number of branches/plant (BP), number of capsules/plant (CP), capsule weight/plant (CWP), seed weight/plant (SWP), husk weight/plant (HW), test weight (TW), number of seeds/capsule (SPC) and oil content (OC). The oil content was measured through minispec time domain-nuclear magnetic resonance (TD-NMR) analyzer (Bruker Corporation, USA).
Genotyping with SSRs
The genomic DNA was isolated from young and freshly harvested leaves of 1 month old plants using DNeasy plant mini kit (Qiagen, Valencia, California) as per manufacturer’s instructions. The quality of DNA was evaluated on 0.8% agarose gel and quantified using Qubit 2.0 fluorometer (Life Technologies, USA) as per standard protocol. Finally, the DNA was diluted to 5 ng/µl for PCR analysis. A set of 50 random SSRs, of which 24 were mapped SSRs from Cloutier et al. (2012a) and rest 26 random SSRs developed from genomic sequences of linseed, were used for genotyping of the 168 accessions. The SSR primers were synthesized with an additional 18 base tag (5′-TGTAAAACGACGGCCAGT-3′) at 5′end to all the forward primers (named as M13 tag) following Schuelke (2000). The PCR was carried out in 10.0 µl reaction volume containing 5 ng of genomic DNA, 1X PCR buffer, 2.5 mM MgCl2, 0.25 mM each dNTPs, 0.5U Taq DNA polymerase (Qiagen), 1.0 pmol forward primer, 3.0 pmol each of both reverse and M13 tag (same as 18 base tag labeled with either 6-FAM, NED, VIC or PET). The PCR amplification was carried out in Veriti Thermal cycler (Applied Biosystems) with an initial denaturation of 5 min at 95 °C followed by 35 cycles of denaturation for 30 s at 94 °C, annealing for 45 s at 48–52 °C (primer specific) and extension for 30 s at 72 °C. Subsequently, 10 additional cycles of denaturation for 30 s at 94 °C, annealing for 45 s at 53 °C, extension for 45 s at 72 °C followed by final extension for 15 min at 72 °C was performed. The PCR amplification was first confirmed on 1.5% agarose gel and then post PCR pool was prepared based on fluorescence labeled primers. For post PCR pooling, 1.0 µl of 6-FAM and 2.0 µl of each VIC, NED and PET labeled PCR product with different SSRs were combined with 13.0 µl of water. Then 1.0 µl of that mix was added to 10.0 µl Hi-Di formamide containing 0.25 µl GeneScan™ 600 LIZ® as internal size standard. This was then denatured for 5 min at 95 °C, quick chilled on ice for 5 min and loaded on ABI 3730xl DNA Analyzer for capillary electrophoresis. The raw data obtained was analyzed by GeneMapper v4.0 software (Applied Biosystems, Foster City, CA, USA) to obtain allele size data.
Statistical analysis
The morphological data of different traits was used to calculate basic statistics and broad sense heritability (h2b) based as described by Allard (1999) using WINDOSTAT software (www.indostat.org). The phenotypic correlation (rp) was calculated as suggested by Johnson et al. (1955). The allelic data of SSRs were subjected to statistical analysis using Power Marker (Liu and Muse 2005) to calculate observed heterozygosity (Ho), gene diversity or expected heterozygosity (He), major allele frequency and polymorphic information content (PIC) value. To understand the genetic relationship pair-wise genetic dissimilarities among all the 168 accessions were calculated according to Jaccard’s coefficient using DARwin 5.0.128 software (Perrier et al. 2003). The calculated dissimilarity matrix was then used to (1) construct a NJ tree with 1000 replicate bootstrap test and (2) perform principal coordinate analysis (PCoA). To determine the number of subpopulation among 168 accessions, Bayesian clustering was carried out using software package STRUCTURE v 2.3.3. The STRUCTURE program was run using admixture model with independent allele frequencies. The presumed population (K) was set from 2 to 15 with five independent runs per K and each run was carried out using 30,000 burn-in period and 100,000 iterations. The optimal value of K was determined by examining Delta K statistic and L (K) according to Evanno et al. (2005) using structure harvester (Earl and Von Holdt 2012). The number of observed alleles (Na), number of effective alleles (Ne), Shannon’s information index (I) and molecular variance (AMOVA) (Excoffier et al. 2009) was calculated with GenAlex 6.5 software.
Association mapping
The association of each marker with different traits was tested using software TASSEL v3.0 (Bradbury et al. 2007). A kinship matrix (K-matrix), the pair-wise relationship matrix which was further used for population correction in the association models was calculated among the accessions with 50 SSRs markers. Two models, (1) GLM adjusted using Q-matrix and (2) the MLM adjusted using both Q-matrix and kinship (K)-matrix was used to test the marker-trait-associations (MTA). The P values were adjusted according to Benjamini and Hochberg (1995) to control the false discovery rate (FDR). These associations were considered with an adjusted P < 0.05.
Results
Phenotypic variability and trait association
The mean and range value of 10 agronomic traits alongwith other statistics are presented in Table 1 while the frequency distributions are shown in Fig. 1. The days of 50% flowering varied from 50.0 to 136.0 with an average of 101.8 ± 19.4. Eighteen accessions were early flowering (<70 days of 50% flowering) while 4 were very late (>130 days). The plant height ranged from 44.33 to 136.8 cm with an average height of 81.0 ± 18.3. Most of the accessions was of average height and only 15 were shorter (<60.0 cm) and 9 were taller (>120.0 cm). The numbers of branches/plant were found variable from 1.6 to 8.0 (average 3.9 ± 1.1), number of capsules/plant varied between 16.1 and 175.0 (average 68.0 ± 33.0). Capsule weight/plant and seed weight/plant ranged from 1.9 to 12.2 g (average 5.9 ± 2.2) and from 1.1 to 9.9 (average 3.9 ± 1.9) respectively. The test weight varied between 2.9 and 11.1 g with an average of 5.8 ± 1.3. The oil content showed lower variability and was found in range of 30.4 to 45.02% with an average of 39.6 ± 2.7%. Only 3 accessions were found to have oil content above 44.0%. The coefficient of variability was found maximum for capsule weight/plant and seed weight/plant (48.5%) while minimum was noticed for oil content (6.8%). The broad sense heritability was found highest for days of 50% flowering (98.4%) followed by number of branches/plant (96.2), test weight (95.0%) and plant height (94.2%). The lowest heritability was noticed for husk weight/plant (38.5%). To understand the relationship among the various morphological and yield related traits, correlation coefficient was also calculated (Table 1). The days of 50% flowering showed significant and negative association with number of capsules/plant (−0.55), capsule weight/plant (0.53), seed weight/plant (−0.62), test weight (−0.53), oil content (−0.39). However, number of branches/plant showed negative but non significant association with most of the traits. Contrary to these, capsule weight/plant and seed weight/plant had positive and significant association plant height (0.35), number of capsules/plant (0.65), test weight (0.49) and oil content (0.25). Oil content had positive and significant association with capsule weight/plant, seed weight/plant and test weight. The frequency distribution plots for these traits (Fig. 1) showed broad Gaussian curves/trends that in most cases were uniformly distributed around the means, consisted with the observed low levels of skewness and kurtosis (Table 1).

Frequency distribution of different agro-morphological traits among 168 linseed accessions
Genotypic variations and genetic distance
The 50 SSRs genotyped across the 168 accessions produces a total of 337.0 alleles with an average of 6.74 alleles per SSRs (Table 2) that ranged from 2 (LU_2155 and LU_3148) to 13 alleles (LU_2332) per SSRs. The maximum number of alleles (13) was shown by SSR LU_2332 followed by SSRs LUSc_196_02, LUSc_38_01 (12 alleles), LU_2651, LU_297, LUSc_160_01 (11 alleles) and LU_283, LUSc_155_02 (10 alleles). The PIC value ranged from 0.19 (LU_197) to 0.87 (LU_2332) with an average of 0.47 ± 0.18. Out of 50, 28 SSRs (56%) showed moderate value of PIC ranging between 0.30 and 0.60 while 12 SSRs (24%) were highly informative having PIC value above 0.61. The rest 20% (10) SSRs had lower PIC value <0.30. The observed heterozygosity ranged from 0.00 (LU_2155, LU_485) to 0.98 (LUSc_27_02) with an average of 0.28 ± 0.25. The 36% (18) SSRs showed observed heterozygosity below 0.10. The gene diversity was in between 0.20 (LU_197) to 0.87 (LU_2332) with an average of 0.51 ± 0.18.
In order to understand the genetic relatedness among the 168 accessions of linseed, the allelic data were used to calculate a pair-wise genetic distance based on Jaccard’s coefficient. The pair wise genetic dissimilarity varied from 0.19 to 0.84 with an average of 0.59 ± 0.07. The maximum genetic dissimilarity of 0.81 was observed between accessions EC 41733 (CG 82) and SJKO-71 (CG 201) followed by 0.80 between CC 12 (CG 40) and RSJ-24 (CG 205) and 0.79 among NP-26 RR SK (CG 148), RJK-14 (CG 215) and Shikha (GP1). The minimum genetic dissimilarity (0.16) was noticed between CG119 and CG125 followed by Kartika (GP28) and Indra Alsi (GP29) (0.23). The minimum, maximum and average genetic dissimilarity of each accession with rest of 167 accessions has also been calculated (Supplementary Table 1). The average genetic dissimilarity of each accession with other accessions varied between 0.51 ± 0.11 (CG125) and 0.68 ± 0.05 (CG138). The minimum was noticed for the accessions CG125, CG119 (0.16) and maximum for accessions CG82, CG201 (0.81). The accessions CI-2067 (CG-52), ILS-153 (CG 119), JABALPUR-367 (CG120) had the lowest range of pairwise genetic dissimilarity coefficient with all other accessions. Considering the genetic distance, the accessions BS-2 (CG31), NP-26 RR SK (CG148), H-8 (CG 108), S-91-45 (CG 186), Parvati (GP-4), GS 234 (GP 53), and JRF 4 (GP 54) were found to be the most divergent among all the accessions studied.
Genetic diversity and population structure
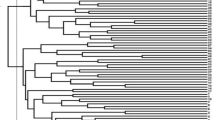
The NJ based dendrogram grouped all the 168 accessions into three major clusters namely cluster I, cluster II, and cluster III (Fig. 2). The cluster I was found to be the largest with 96 accessions (57%) followed by the cluster III having 51 accessions (30%). The clusters II had 21 accessions (13%). The cluster I consisted of accessions all from core germplasm category while the cluster III had all the accessions from germplasm category except one (CG212). The cluster II was found to have mixed accessions from both the categories i.e. 13 from core germplasm and 8 from germplasm. The genetic diversity among accessions was also confirmed by scatter plot derived through PCoA (Fig. 3). The first three coordinate represents 23.11% of the total variation, with the first coordinate explain 15.56%, the second coordinate 3.85% and third coordinate 3.70%. Similar to NJ clustering 96 accessions of core-germplasm clustered together at left portion of the plot while 58 accessions of germplasm and 14 accessions of core-germplasm grouped at right portion of the plot. The accession CG138 was sparsely situated.

Dendrogram of 168 linseed accessions obtained from 50 SSR marker based on Jaccards coeffficient using NJ clustering method

2-D scatter plot of principal coordinate analysis (PCoA) of 168 linseed accession based on 50 SSR data
The model based simulation of population structure was analyzed to investigate the subpopulations among the 168 accessions varying from K = 2 to K = 10. The optimum number of subpopulation was determined by the maximum ΔK value obtained through structure harvester. The K = 4 was found to be the most appropriate subpopulations (Supplementary Fig. 1a, b). The bar plot of model based clustering showed that 95 accessions (56.6%) were assigned to subpopulation 1 including only core-germplasm. The subpopulation 4 comprised of 46 accessions (27.4%) with 91.0% germplasm and 9.0% core-germplasm accessions (Fig. 4). The subpopulation 2 had 12 accessions (7.14%) with 83.3% germplasm and 16.7% core-germplasm. The subpopulation 3 had 15 (8.93%) accessions with 60.0% germplasm and 40.0% core-germplasm. Similar to the NJ clustering the structure analysis also differentiated germplasm, core-germplasm accessions and mixed accessions. The clusters of NJ dendrogram and subpopulation derived from structure analysis showed maximum similarity for accessions compositions. In subpopulation 1 and cluster I both have only core-germplasm, the subpopulation II and III both have admixture of core-germplasm similar to cluster II of NJ dendrogram. The cluster III mostly contains germplasm, as subpopulation IV.

Model-based Bayesian clustering performed in STRUCTURE for K = 4 populations of 168 genotypes of linseed
Based on NJ clustering, AMOVA was carried out considering 3 populations to test the significance of genetic structure. It was noticed that 79% of the total genetic variation was within the population and 21% was among the population (Table 3). The average pairwise ΦPT (similar to FST) was 0.21 supported the strong population structure. The mean of Nei genetic diversity, effective number of alleles and shannon’s information index were estimated as 0.14 ± 0.005, 1.22 ± 0.009 and 0.23 ± 0.007 respectively (Table 4).
Association analysis
Association analysis was performed for all the 10 traits in 168 accessions. We have tested two model i.e. GLM-Q model and MLM-Q + K model to determine the association and data are presented in Table 5. The GLM approach detected large number of association between the markers and traits which reduces after FDR corrections for multiple testing. With no FDR correction, the GLM detects 4 markers each associated with days to 50% flowering, plant height, branches/plant, 3 markers each for seed weight/plant, seeds/capsules, 2 markers each for number of capsules/plant, capsule weight/plant, and 1 for husk weight/plant and test weight. After FDR correction, only 1 marker (LU_3043) for days to 50% flowering, 2 markers (LUSc_66_01 M31, LU_2332) for plant height, 1 marker (LU_2332) for seeds/capsules and 1 marker (LU_197) for test weight were found to be associated (corrected P < 0.05). Further, the MLM (Q + K model) approach detected several markers associated with different traits and many of them were common with GLM and few were new markers. Out of several associated markers, only 1 marker (LU_3043) was found to be truly associated with days to 50% flowering as it showed significant corrected P value (P < 0.005). The marker LU_3043 showed consistent association with days to 50% flowering in both GLM and MLM analysis.
Discussion
The knowledge of genetic diversity in linseed makes it possible to adopt appropriate breeding strategies for developing new and high yielding varieties to meet out the both human and industrial demands. The genetic improvement is a continuous and ongoing process, therefore the creation and knowledge of genetic variability is an important step to search new sources of variation for further genetic improvement. Hence, evaluating different sets of genetic materials with appropriate tools would be of useful for identifying diverse accessions/genotypes to be incorporated in different breeding programs.
In case of linseed different types of DNA based markers including AFLP, RAPD, retrotransposon, ISSR, SSRs, SNP etc. have been used for evaluating the genetic diversity and linkage mapping (Cloutier et al. 2010; Oh et al. 2000; Spielmeyer et al. 1998). Over other types of DNA markers, SSRs are widely used for genetic diversity and structure analysis in several crop plants (Cheng et al. 2015; Yan et al. 2009; Cane et al. 2014; Cui et al. 2013). In the present investigation, we applied 50 widely distributed SSRs to evaluate the genetic diversity and identify diverse accessions/group of accessions among a large set of 168 accessions of linseed. A total of 337 alleles were detected from 50 SSRs with an average of 6.74 alleles per SSRs. This average allele number was found to be higher than that reported earlier (Roose-Amsaleg et al. 2006; Deng et al. 2011; Pali et al. 2014; Soto-Cerda et al. 2012, 2013; Cloutier et al. 2012b). The higher allele number might be due to the large and diverse set of accessions and widely distributed SSRs used in the present study. The three markers namely LU_2332, LUSc_38_01 and LuSc_196_02 having 13, 12 and 12 alleles with PIC value above 0.80 seems to be highly informative and could be further utilized in evaluating other sets of linseed germplasm. The average PIC value (0.47) was found to be higher than those reported by Soto-Cerda et al. (2011, 2012, 2013), Cloutier et al. (2009, 2012b). While Deng et al. (2010) reported slightly higher average PIC value (0.56) than found in the present study. Among the other studies with Indian linseed, Rajwade et al. (2010) studied the genetic diversity and observed lower genetic diversity (0.15) and PIC (0.18) value compared with our study. Bickel et al. (2011) observed similar average PIC value (0.47) using 42 polymorphic SSRs primers. The polymorphism level can vary significantly across the studies depending upon the number of accessions surveyed and nature of genetic materials. Our result show comparatively higher level of genetic diversity and PIC value as compared to previous studies where low level of diversity reported (Rajwade et al. 2010; Soto-Cerda et al. 2012; Cloutier et al. 2012b).
In this study, 168 accessions of linseed show high level of genetic diversity (average dissimilarity coefficient 0.59 ± 0.07). Eleven accessions namely EC 41733 (CG 82), SJKO-71 (CG 201), CC 12 (CG 40), RSJ-24 (CG 205, BS-2 (CG31), NP-26 RR SK (CG148), H-8 (CG 108), S-91-45 (CG 186), Parvati (GP-4), GS 234 (GP 53), JRF 4 (GP 54) were highly diverse among all the accessions studied. The genetic differentiation among isolated clusters based on Nei’s genetic diversity was observed (0.14) and Shannon’s diversity index (0.23) was low compared to Kale et al. (2012).
To understand the level of genetic divergence and population structure NJ based dendrogram and model based structure plot was generated. The accessions belong to subpopulation 1 and cluster I showed maximum similarity except one genotype CG 138. The subpopulation 2 and 3 showed admixture type similar to cluster II. The subpopulation 2 and 3 have 27genotype and cluster II have 21 accessions and 17 accessions were common in both. The subpopulation 4 shows mostly germplasm accessions as same to the cluster III. The NJ dendrogram and population structure analysis produces almost similar pattern of grouping of accessions indicating that any one of the method could be adopted to decipher genetic stratification and identification of diverse accessions of linseed. The clustering derived from PCoA corresponds to the clustering of both NJ and structure analysis and revealed the robustness of the markers used in the present investigation and confidence of statistical analysis. Further, the AMOVA analysis show significance of the genetic structure, indicated 79% of the genetic variation resided within the populations and 21% was due to differences among the three population identified based on NJ analysis. In AMOVA the FST value was observed 0.21 supporting strong population structures. Our results were found to be similar to those reported earlier (Soto-Cerda et al. 2012). The high broad sense heritability of days to 50% flowering and plant height and association among these traits suggests that breeding for early flowering and shorter plant type is feasible. Further, the correlation of seed weight/plant with number of capsules/plant, capsule weight/plant and test weight indicated that indirect selection through these traits might be helpful in developing high yielding linseed varieties. The model based structure analysis characterized the accessions in subpopulations and was useful for association mapping (Newell et al. 2010) and further breeding strategies.
Association mapping is widely being utilized for detecting marker-trait association based on the genetic resources routinely used for genetic improvement programs (Font I Forcada et al. 2015; Cheng et al. 2015; Yan et al. 2009; Cai et al. 2013). As compared to bi-parental QTL mapping, association mapping increases the range of natural variation that can be exploited in a single experiment and potential genomic regions could be identified (Jin et al. 2010). However, this method has chances of false positives mainly due to the population structure (Pritchard et al. 2000) and familial relatedness (Yu et al. 2006). Several linear and mixed models have been proposed to correct the population structure and reduce the false positive factors (Pritchard et al. 2000; Price et al. 2006; Yu et al. 2006). Association mapping in linseed have been carried out for various agronomic and seed quality traits (Soto-Cerda et al. 2013, 2014) using SSR markers. Soto-Cerda et al. (2013) identified 12 QTLs for six agronomic traits while 9 QTLs were identified for seed quality traits (Soto-Cerda et al. 2014). In the present investigation only 1 QTL was potentially identified for days to 50% flowering. One SSR Lu_3043 was consistently associated with days to 50% flowering by both GLM and MLM marker trait association analysis. These results suggest the usefulness of this marker for further characterization of this trait. The poor association was due to less number of markers and or accessions used. In order to tap maximum number of QTLs for important agronomic traits diverse set of accessions and large number of markers should be used.
Conclusion
The present investigation added knowledge on genetic variation, population structure and genetic diversity status of 168 linseed accessions. Several important accessions have been identified which could be of potential for future linseed genetic improvement program. Additionally, one important QTL related to flowering trait have also been identified.
References
Allaby RG, Peterson GW, Merriwether DA, Fu YB (2005) Evidence of the domestication history of flax (Linum usitatissimum L.) from genetic diversity of the sad2 locus. Theor Appl Genet 112:58–65
Allard RW (1999) Principles of plant breeding, 2nd edn. Wiley, New York
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Bickel CL, Gadani S, Lukacs M, Cullis CA (2011) SSR markers developed for genetic mapping in flax (Linum usitatissimum L.). Res Rep Biol 2011:23–29
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Cai S, Yu G, Chen X, Huang Y, Jiang X, Zhang G, Jin X (2013) Grain protein content variation and its association analysis in barley. BMC Plant Biol 13:35
Cane MA, Maccaferri M, Nazemi G, Salvi S, Francia R, Colalongo C, Tuberosa R (2014) Association mapping for root architectural traits in durum wheat seedlings as related to agronomic performance. Mol Breed 34:1629–1645
Chandrawati, Maurya R, Singh PK, Ranade SA, Yadav HK (2014) Diversity analysis in Indian genotypes of linseed (Linum usitatissimum L.) using AFLP marker. Gene 549:171–178
Cheng P, William H, Yu M, Coyne CJ, Mazourek M, Grusak MA, Sam F, McGee RJ (2015) Association mapping of agronomic and quality traits in USDA pea single-plant collection. Mol Breed 35:75
Cloutier S, Niu Z, Datla R, Duguid S (2009) Development and analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor Appl Genet 119:53–63
Cloutier S, Ragupathy R, Niu Z, Duguid S (2010) SSR- based linkage map of flax (Linum usitatissimum L.) and mapping of QTLs underlying fatty acid composition traits. Mol Breed 28:437–451
Cloutier S, Ragupathy R, Miranda E, Radovanovic N, Reimer E, Walichnowski A, Ward K, Rowland G, Duguid S, Banik M (2012a) Integrated consensus genetic and physical maps of flax (Linum usitatissimum L.). Theor Appl Genet 125:1783–1795
Cloutier S, Miranda E, Ward K, Radovanovic N, Reimer E, Walichnowski A, Datla R, Rowland G, Duguid S, Ragupathy R (2012b) Simple sequence repeat marker development from bacterial artificial chromosome end sequences and expressed sequence tags of flax (Linum usitatissimum L.). Theor Appl Genet 125:685–694
Cui D, Xu CY, Tang CF, Yang CG, Yu TQ, Xin-xiang A, Cao GL, Xu FR, Zhang JG, Han LZ (2013) Genetic structure and association mapping of cold tolerance in improved japonica rice germplasm at the booting stage. Euphytica 193:369–382
Deng X, Long SH, He DF, Li X, Wang YF, Liu J, Chen XB (2010) Development and characterization of polymorphic microsatellite markers in Linum usitatissmum. J Plant Res 123:119–123
Deng X, Long S, He D, Li X, Wang Y, Hao D, Qiu C, Chen X (2011) Isolation and characterization of polymorphic microsatellite markers from flax (Linum usitatissimum L.). Afr J Biotechnol 10:734–739
Diederichsen A, Ulrich A (2009) Variability in stem fibre content and its association with other characteristics in 1177flax (Linum usitatissimum L.) genebank accessions. Ind Crop Prod 30:33–39
Earl DA, Von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evano method. Conserv Genet Resour 4:359–366
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Hofer T, Foll M (2009) Detecting loci under selection in a hierarchically structured population. Heredity 103:285–298
Flint-Garcia SA, Thuillet AC, Yu J, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman M, Buckler E (2005) Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44:1054–1064
Font I Forcada C, Velasco L, Company IRS, Martí IÁF (2015) Association mapping for kernel phytosterol content in almond. Front Plant Sci 6:530
Green A, Chen Y, Singh S, Dribnenki P (2008) Flax. In: Kole C, Hall TC (eds) A compendium of transgenic crop plants. Blackwell, Oxford, pp 199–206
Hickey M (1988) 100 families of flowering plants, 2nd edn. University Press, Cambridge
Jin L, Lu Y, Xiao P, Sun M, Corke H (2010) Genetic diversity and population structure of a diverse set of rice germplasm for association mapping. Theor Appl Genet 121:475–487
Johnson HW, Robinson HF, Comstock RE (1955) Estimates of genetic and environmental variability in Soybean. Agron J 47:314–318
Kale SM, Pardeshi VC, Kadoo NY, Ghorpade PB, Jana MM, Gupta VS (2012) Development of genomic simple sequence repeat markers for linseed using next-generation sequencing technology. Mol Breed 30:597–606
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Newell MA, Cook D, Tinker NA, Jannink JL (2010) Population structure and linkage disequilibrium in oat (Avena sativa L.): implications for genome-wide association studies. Theor Appl Genet 122:623–632
Oh TJ, Gorman M, Cullis CA (2000) RFLP and RAPD mapping in flax (Linum usitatissimum). Theor Appl Genet 101:590–593
Pali V, Verma SK, Xalxo MS, Saxena RR, Mehta N, Verulkar SB (2014) Identification of microsatellite markers for fingerprinting popular Indian flax (Linum usitatissimum L.) cultivars and their utilization in seed genetic purity assessments. Aust J Crop Sci 8:119–126
Pali V, Mehta N, Verulkar SB, Xalxo MS, Saxena RR (2015) Molecular diversity in Flax (Linum usitatissimum L.) as revealed by DNA based markers. Int J Plant Res 28:157–165
Perrier X, Flori A, Bonnot F (2003) Data analysis methods. In: Hamon P, Seguin M, Perrier X, Glaszmann JC (eds) Genetic diversity of cultivated tropical plants. Enfield Science Publishers, Montpellier, pp 43–76
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genet 155:945–959
Rajwade AV, Arora RS, Kadoo NY, Harsulkar AM, Ghorpade PB, Gupta VS (2010) Relatedness of Indian flax genotypes (Linum usitatissimum L.): an inter-simple sequence repeat (ISSR) primer assay. Mol Biotechnol 45:161–170
Roose-Amsaleg C, Cariou Pham E, Vautrin D, Tavernier R, Solignac M (2006) Polymorphic microsatellite loci in Linum usitatissimum. Mol Ecol Notes 6:796–799
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Soto-Cerda BJ, Carrasco RA, Aravena GA, Urbina HA, Navarro CS (2011) Identifying novel polymorphic microsatellites from cultivated flax (Linum usitatissimum L.) following data mining. Plant Mol Biol Rep 29:753–759
Soto-Cerda BJ, Maureira-Butler I, Munoz G, Rupayan A, Cloutier S (2012) SSR-based population structure, molecular diversity and linkage disequilibrium analysis of a collection of flax (Linum usitatissimum L.) varying for mucilage seed-coat content. Mol Breed 30:875–888
Soto-Cerda BJ, Diederichsen A, Ragupathy R, Cloutier S (2013) Genetic characterization of a core collection of flax (Linum usitatissimum L.) suitable for association mapping studies and evidence of divergent selection between fiber and linseed types. BMC Plant Biol 13:78
Soto-Cerda BJ, Duguid S, Booker H, Rowland G, Diederichsen A, Cloutier S (2014) Association mapping of seed quality traits using the Canadian flax (Linum usitatissimum L.) core collection. Theor Appl Genet 127:881–896
Spielmeyer W, Green AG, Bittisnich D, Mendham N, Lagudah ES (1998) Identification of quantitative trait loci contributing to Fusarium wilt resistance on an AFLP linkage map of flax (Linum usitatissimum). Theor Appl Genet 97:633–664
Vavilov N (1926) Studies on the origin of cultivated plants, vol 16. Bull Appl Bot Plant Breed, Leningrad, USSR, pp 139-248
Westcott NA, Muir AD (2003) Flax seed lignan in disease prevention and health promotion. Phytochem Rev 2:401–417
Yan WG, Li Y, Agrama HA, Luo D, Gao F, Lu X, Ren G (2009) Association mapping of stigma and spikelet characteristics in rice (Oryza sativa L.). Mol Breed 24:277–292
Yu JM, Pressoir G, Briggs WH, Bi IV, Yamasaki M (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zeist WV, Bakker-Heeres JAH (1975) Evidence for linseed cultivation before 6000 BC. J Archeol Sci 2(3):215–219
Zohary D, Hopf M (2000) Domestication of plants in the old world: the origin and spread of cultivated plants in West Asia, Europe and the Nile Valley. Oxford University Press, Oxford, p 316
Acknowledgements
Authors thank the Director, CSIR-NBRI, Lucknow for providing the facilities to carry out the present investigation. Financial support in form of DST-INSPIRE Fellowship to Chandrawati is gratefully acknowledged.
Author’s contribution
HKY, SAR, RK and SK conceived and designed the research. C, NS, carried out the experiments. PKS provided genetic materials. HKY, VKY, SAR and RK analyzed data. C, NS, SK and HKY wrote the manuscript. All authors read and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chandrawati, Singh, N., Kumar, R. et al. Genetic diversity, population structure and association analysis in linseed (Linum usitatissimum L.). Physiol Mol Biol Plants 23, 207–219 (2017). https://doi.org/10.1007/s12298-016-0408-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-016-0408-5