
Abstract
Prostate cancer is the World's second most frequent malignancy, with the fifth-highest male mortality rate. In advanced prostate cancer patients, point mutations such as T877A and W741L are prevalent, imparting treatment resistance and hence promoting cancer development. The emergence of drug resistance in prostate cancer necessitates the development of suitable ligands to allow for stronger interactions with the receptors, which can inhibit cancer progression. The present study focuses on flavonoids produced by plants, which may act as inhibitors of point mutations like T877A and W741L in prostate cancer. This research was conducted using an in-silico method where the compound Glabranin and its derivatives were virtually screened to identify potential drugs for combating such point mutations. Thirty-five Molecular Dockings were performed to find the ligand-receptor complexes with the lowest binding energy. Moreover, employing a variety of tools, ligands were evaluated for drug-likeness and toxicity, indicating a promising drug candidate. Based on the results of Molecular Docking, Drug-likeness, and ADMET testing, eight structures were subjected to a 100 ns Molecular Dynamics simulation. A QSAR analysis was also performed based on the simulation findings. In this study, it was revealed that GlaMod2 phytocompound was effective against T877A and W741L mutations in prostate cancer. It was observed that the phytocompound was stable and had potential properties for the development of a novel drug to combat prostate cancer and drug resistance This phytocompound may therefore be effective in the development of prostate cancer inhibitors for patients with mutant androgen receptors.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glabranin is a flavanone, a subclass of the flavonoid group. These compounds are derived from the leaves of the Glycyrrhiza glabra L. plant. This plant extract is believed to be used to cure coughs, catarrhal infections, and irritated mucous membranes of the urinary organs, as well as throat lozenges, pastilles, stomach ulcers, Addison's disease, and a variety of other ailments [1,2,3]. This plant has also been shown to have antimicrobial and anti-arthritic properties [4]. Glabranin-A and Glabranin-B are saponins produced by the roots of the Glycyrrhiza glabra L. plant. These saponins are glycyrrhetic acid glycosides that are extremely important in biological areas [5, 6]. The entire plant of Glycyrrhiza glabra has powerful antioxidant, antiulcer, anticancer, antimutagenic, anti-HIV, and hepatoprotective properties [7, 8]. It has been discovered that Licochalcone A, extracted from the roots of Glycyrrhiza glabra, has an anticancer effect against breast cancer, lung cancer, stomach cancer, liver cancer, colon cancer, kidney cancer, and leukemia. An additional file shows the structure of the Glabranin Compound (Fig. S1).
Prostate cancer originates in the prostate gland in which cells of the gland mutate into malignant cells and spreads to other areas of the body, notably to the bones and lymph nodes. Dyspnoea, weight loss, spinal cord compression, fever, vertigo, urine urgency, sexual dysfunction, nocturia, fatigue, chronic pain, and urinary tract infection are common symptoms in individuals with prostate cancer [9, 10]. The progression of prostate cancer is aided by mutations in the p53 gene and the resulting malfunctioning of the cell cycle, hormone signaling, and apoptosis signaling pathways [11, 12]. Along with p53, androgen receptor (AR) plays an important role in the occurrence and/or relapse of prostate cancer by causing abnormalities in androgen signaling (Fig. S1) and several distinct somatic AR mutations have been identified in prostate cancer patients [13]. By reducing/losing the selectivity for the antagonists, point mutations can activate AR. For example, Thr877 and Trp741 in the AR's ligand-binding domain are frequently found to be associated with mutations in relapsed prostate cancer patients [14,15,16]. Point mutations like T877A and W741L in prostate cancer convert the antagonist into an agonist, allowing cancer cells to grow more quickly. Prostate cancer drugs such as Bicalutamide and Flutamide, rather than inhibiting, stimulate the transcription role of T877A-mutated AR and W741L-mutated AR as a result of the substrate-binding site alteration [17]. Consequently, designing new ligands that are more compliant with receptors is expected to tackle drug resistance. Because AR plays such a significant role in prostate cancer, understanding the molecular mechanism underlying the malignancies controlled by AR will offer additional knowledge needed for developing inhibitors against T877A and W741L mutations in prostate cancer (see Supplementary Information, Fig. S2).
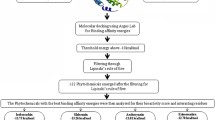
A molecular docking study was conducted against 803 phytochemicals in 2017, revealing that the phytocompound Glabranin may be one of the potential treatment alternatives against wild androgen receptors [18]. Nonetheless, the study was limited to molecular docking of the phytocompounds at natural androgen receptors and did not include Glabranin testing against mutant AR, and no molecular modeling studies were conducted to assess the compound's stability in the complex form. This widens the scope of the investigation into Glabranin and its derivatives against mutations in prostate cancer. As a consequence, the extent of use of Glabranin and its derivatives against androgen receptors with T877A and W741L mutations was investigated in this research. The current study employed an in-silico technique to identify the potentiality of Glabranin and its derivatives as a potent blocking agent against T877A and W741L mutations of AR. The study involved the use of molecular docking as well as drug likelihood and toxicity check tools along with molecular dynamics simulation to investigate the ligand-receptor complex’s stability and QSAR (Quantitative structure–activity relationship) prediction to predict the inhibitory action of the potential candidate [19,20,21].
Methods
Structure Preparation
AR structures were obtained from PDB (Protein Data Bank) for molecular docking studies. This study included four types of AR: wild type (PDB ID-1E3G), AR with T877A mutation (PDB ID-2AX6), AR with W741L mutation (PDB ID-1Z95), and AR with both T877A and W741L mutations generated using Chimera software [22,23,24]. The normal ligands of both receptors, namely Hydroxyflutamide from the 2AX6 target protein and R-bicalutamide from the 1Z95 target protein, were removed using Chimera software [25]. The WHAT IF Interface, was used to detect and simulate the missing residues in the receptor PDB archives. The energy of the receptors was also reduced using the Programme Chimera.
Glabranin, a flavonoid phytocompound, its four derivatives, Hydroxyflutamide and R-bicalutamide (pre-existing ligands), and two commercially available drugs, Bicalutamide and Flutamide, have been used as ligands in this study [26,27,28,29,30]. The ligands canonical smiles were acquired from the PubChem database. Chimera software was used to build the ligand structures. The derivatives of the Glabranin compound were obtained using the ChemAxon Programme [31]. In this analysis, Bicalutamide and Flutamide, which are commercially available drugs for the treatment of prostate cancer, were also docked against the AR. The Protein Data Bank (PDB) was employed in this study to classify the AR’s active binding site. The ligands were subjected to energy minimization using Chimera software to escape any irregular geometries and to attain a low energy conformation [32]. 5000 Steepest Descent and 5000 Conjugate Gradient cycles were used to accomplish this energy minimization.
Molecular Docking
The role of the point mutation in transforming an AR antagonist into an agonist was explored using docking analysis between the androgen receptor and ligands. Autodock Software was used to perform 35 molecular dockings (blind and site-specific docking). The non-polar hydrogen atoms were excluded from the receptor archives, and partial charges were added. Gasteiger partial charges were allocated to both ligands and non-polar hydrogen atoms were combined, while Kollman charges and polar hydrogen atoms were assigned to receptors using ADT (Autodock Tools) [33]. PDB (Protein Data Bank) was used for determining the AR's catalytic sites for site-specific dockings. The AutoGrid Software was used to create a grid box with numerous points in xyz of 22 × 20 × 20 for the 1E3G receptor, 24 × 22 × 24 for the 2AX6 receptor, 26 × 26 × 26 for the 1Z95 receptor, and 22 × 20 × 20 for the receptor with both mutations in the ideal conformational state of docking. Blind docking was performed using the Autodock software to validate the correctness of the ligands binding to their specific positions as predicted by the PDB protein structures. For all molecular dockings, a grid box of 1.000 Å was employed. Following the completion of molecular docking, PyRx (a Graphical User Interface for the Autodock tool that does virtual screening) was used to carefully analyze the binding energies of the twenty best postures from each protein–ligand complex [34, 35]. The LigPlotv.1.4.5 computer program was used to analyze and display the molecular docking results of protein–ligand complexes [36, 37].
Physiochemical Property Prediction
The canonical smiles of the ligands were obtained from the PubChem server. These compound smiles were entered into Molinspiration online software, pkCSM Server (Predicting Small-Molecule Pharmacokinetic Properties Using Graph-Based Signatures), and free online Lipinski Filters server to calculate Lipinski's Rule, also known as the Rule of Five [38]. According to this Rule of Five (ROF), the compound's Molecular mass must not surpass 500 Dalton, the LogP partition coefficient must not surpass five, the number of hydrogen bond donors must not surpass five, and the number of hydrogen bond acceptors must not surpass ten. In addition to Lipinski's Rule, other drug-likeness rules were evaluated using the SwissADME server and DruLiTo Software [39]. Furthermore, the compounds' canonical smiles were submitted to pkCSM, Pro Tox-II server, admetSAR server, and ADMETlab 2.0 online server to analyze ADMET characteristics. Using LigPlot + version 2.2 software, the most favorable binding conformations of the various complexes were chosen and tested for hydrogen bond interactions with the protein [40].
Molecular Dynamics Simulation of Protein–Ligand Complex
The trajectories of the selected complexes and the receptors were analyzed through 100 ns of MD (Molecular Dynamics) simulation using GROMACS (GROningen MAchine for Chemical Simulations) 4.6.2 software package under AMBER99SB force field (Assisted Model Building with Energy Refinement) [41]. The ligand topology files were created using ACPYPE (Ante Chamber PYthon Parser interface) software. The temperature was set to 310 K, and the systems were equilibrated for 200 ps in the NVT ensemble (Number of particles, Volume, and Temperature) and another 200 ps in the NPT ensemble (Number of particles, Pressure, and Temperature). The systems were subjected to a production MD run in an NPT ensemble for 100 ns after heating and equilibration. A total of seven models were examined. The MD trajectories were analyzed using the GROMACS software. Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and Radius of Gyration (Rg) were used to evaluate the compound trajectories. The visualization program VMD (Visual Molecular Dynamics) was used, and the diagrams were plotted using QtGrace plotting techniques.
MM-PBSA Calculations
The free energy measurements of the complexes were conducted using the Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) method after the basic analysis of MD simulations [42]. (∆Gbind = ∆EMM + ∆Gsolv − T∆S) according to MM/PBSA, where ∆EMM is the sum of internal, van der Waals, and electrostatic energy, T∆S is the conformational energy, and ∆Gsolv is the polar and nonpolar solvation energy [43]. The g_mmpbsa tool was used to quantify three energy components: potential energy in a vacuum, polar solvation energy, and non-polar solvation energy. A python script called MmPbSaStat.py from the g_mmpbsa package was used to predict average binding energies. QtGrace was used to plot both of these files.
QSAR Prediction
The QSAR study was first conducted on 30 identified prostate cancer drug targets. These 30 drug target compounds’ IC50 values were obtained from the ChEMBL database [44]. EasyQSAR, a freeware software, was used to perform Multiple Linear Regression Analysis on these 30 identified prostate cancer drug targets (24 training sets and 6 test sets) to derive the QSAR equation. MarvinSketch 19.7 was used to measure seven molecular descriptors: Molecular weight, LogP, Polarizability, Refractivity, Molecular Surface area, Polar Surface Area, and Hydrophilic-Lipophilic Balance (HLB) [45]. The QSAR equation was validated using six known drug targets for prostate cancer. Using EasyQSAR software, this model was then used to predict the IC50 value of the selected phytocompound [46].
Results
Molecular Docking
Molecular docking research was conducted to determine the effectiveness of possible ligands against wild-type AR and mutated AR. In the present sample, four distinct types of ARs were picked, i.e., wild-type AR (PDB ID: 1E3G), T877A mutated AR (PDB ID: 2AX6), W741L mutated AR (PDB ID: 1Z95), and both mutated AR generated by UCSF Chimera. The phytocompound Glabranin was used for docking tests along with its 4 more derivatives and the drugs commonly used in prostate cancer, Bicalutamide and Flutamide, were used as a standard.
Initially, Glabranin, Bicalutamide, and Flutamide were docked against Wild Type AR, the PDB-ID of which is 1E3G. The binding energy of Glabranin to the wild AR (1E3G) was observed to be − 7.9 (kJ/mol) while that of Bicalutamide and Flutamide against wild AR was observed to be − 4.46 and − 6.47 kcal/mol respectively. An additional file shows the Molecular Docking of Glabranin, Bicalutamide, and Flutamide against Wild AR in Tabular Form (See Supplementary Information, Table S1). It was observed from Table S1 that the compound Glabranin had greater binding in comparison to the commercially available drugs. The docking effects of these three compounds along with their canonical smiles are depicted in Table S1 as supplemental.
Following that, docking studies were performed with pre-existing ligands to confirm the Glabranin compound's potential against T877A and W741L mutations (Hydroxyflutamide against 2AX6 with T877A mutation and R-bicalutamide against 1Z95 with W741L mutation). The binding energy levels of Hydroxyflutamide (pre-existing), Bicalutamide and Flutamide drug (controls), and compound Glabranin against 2AX6 receptor (with T877A mutation) were observed to be − 6.62, − 6.4, − 5.71, − 7.37 kcal/mol, respectively. An additional file shows the Molecular Docking of Androgen Receptor against pre-existing, controls and ligand candidates in Tabular Form (See Online resource, Table S2). It was observed that against the 2AX6 receptor (with T877A mutation) compound Glabranin in comparison to commercially available drugs and pre-existing ligands showed a better binding affinity with the binding energy of − 7.37 kcal/mol. Similarly, the binding energy levels of R-bicalutamide (pre-existing), Bicalutamide and Flutamide drug (controls), and compound Glabranin against 1Z95 receptor (with W741L mutation) were observed to be − 7.91, − 3.31, − 4.93, − 6.96 kcal/mol, respectively (Table S2). In comparison to the commercially available drugs (controls), the compound Glabranin showed a good binding affinity with a binding energy of -6.96 kcal/mol. It was also observed that Glabranin compounds binding energy was close to R-bicalutamide (pre-existing ligand) with a binding energy of − 7.91 kcal/mol (Table S2).
In addition to the early results stated above, four Glabranin derivatives were docked against 2AX6 (T877A mutant AR), 1Z95 (W741L mutant AR), and BothMut receptors with both mutations T877A and W741L, as shown in Table 1. To validate the docking results, Autodock software was employed to perform both site-specific docking and blind docking. Mutant androgen receptors with Glabranin derivatives showed greater binding energies than Bicalutamide and Flutamide, which were employed as controls, according to site-specific docking (refer to Table S2). Their inhibition constant was also calculated in addition to their binding energy (Table 1). The lower the inhibition constant (µM), the higher the binding affinity and the less drug needed to block the activity of the mutant androgen receptor. Because the findings of blind docking were inadequate in contrast to the results of site-specific docking, the binding energy, and inhibition constant were calculated in site-specific docking to appropriately assess the results, as shown in Table 1. Here, Table 1 summarizes all molecular docking scores of Glabranin derivatives against mutated ARs.
Docking studies have shown that the mutant Androgen receptors with the derivatives of the Glabranin compound have 12 docking complexes with improved binding energy. When blind docking findings were compared to site-specific docking results, the former was revealed to have considerably greater binding energy, indicating that docking integrating the expected catalytic/binding site improved the precision and efficiency of the Androgen Receptor-Ligand binding. Following a comparison of site-specific binding energy and inhibition constants, it was revealed that compounds with high binding energy had good inhibition constant values, showing a proportional connection between binding energy and inhibition constant.
Drug-Likeness Screening
On screening the compound Glabranin and its derivatives, it was observed that four of them, viz. Gla, GlaMod1, GlaMod2, and GlaMod3 satisfied Lipinski’s rule of five and other rules of drug-likeness. This screening is critical for accelerating the production of a compound as a possible drug or drug target [47,48,49,50,51]. The presence of functional groups, ring systems, and strong atom proportion are all closely linked to drug-likeness and physiochemical properties, so certain derivatives that broke Lipinski's law may be attributed to them. Since one of the most important screening tests for a possible medication is drug-likeness, only compounds that followed Lipinski's Rule of Five and other rules were subjected to the ADMET test. The compound GlaMod4 could not qualify for the drug likeliness screening. Hence, this compound was not considered for further analysis. The sampling of all compounds/ligands for drug likeliness potential using several tools has been summarized in Table 2.
ADMET Screening
The compounds were tested for toxicity using the pkCSM web server, Pro Tox-II (Toxicity) server, and admetSAR (Toxicity). These servers help in the screening of several pharmacokinetic properties (ADMET) of the medications, which include absorption, distribution, metabolism, excretion, and toxicity. These servers may also test for carcinogenicity, hepatotoxicity, and skin sensitivity. The parameters included in this study under Absorption were P-Glycoprotein Substrate, P-Glycoprotein-1, and 2 inhibitors; the parameters included under Distribution were Blood–Brain Barrier Permeability (BBB) whose logBB > 0.3, Central Nervous System Permeability (CNS) whose log PS > -2; the parameters included under Metabolism were Cytochrome P450 inhibitors (CYP2D6, CYP3A4) and Cytochrome P450 substrate When the compounds Glabranin and GlaMod1, GlaMod3 were tested for the ADMET test using the pkCSM server, they were shown to be mutagenic. Using the Pro Tox-II (Toxicity) service, the toxicity of all four compounds was predicted to be inactive. GlaMod2 compound was discovered to have drug-like potential and passed the toxicity test utilizing several methods. The selected compounds were screened based on their toxicity level, as well as other pharmacokinetic properties. The compounds Gla, GlaMod1, GlaMod3, and GladMod4 were seen to be mutagenic in an ADMET test. Overall, it was observed that out of 4 compounds that showed drug-likeliness capacity, only compound GlaMod2 passed the toxicity check. Since these compounds were found to be non-toxic, they could be used in in-vitro experiments. The findings of the toxicity test have been summarized in Table 3.
The final complexes selected for Molecular Dynamic Simulation were 2AX6 + GlaMod2 complex, 1Z95 + GlaMod2 complex, and BOTHUT + GLAMOD2 complex, based on the Molecular Docking Study, Drug Likeliness Capacity (Lipinski's Rule of Five), and Toxicity Check (ADMET). The binding patterns of the molecular docking findings of these final selected complexes were examined using LigPlot Software. An additional file shows the binding pattern of the selected docking complexes (See Supplementary Information, Fig. S3).
Molecular Dynamics Simulation
After molecular docking and screening of compounds for Drug Likeliness and ADMET evaluation, the Molecular Dynamics Simulations technique was used to determine the stability of the complexes in this analysis (Table 3). This method was employed in this investigation to test the long-term stability of the complexes. The compounds in complex with mutated AR that demonstrated strong binding energy and passed both the Drug Likeliness and the ADMET tests were chosen for Molecular Dynamics Simulations in this analysis. Using the package GROMACS 4.6.2, this was done to expose the flexibility and overall stability of the chosen docked complexes at 100 ns. QtGrace software was used to calculate the variance and fluctuation in the complexes using RMSD, RMSF, and Rg [52, 53]. The following is an analysis of the effects of Molecular Dynamics Simulations:
Root Mean Square Deviation (RMSD)
Throughout the 100 ns MD simulation, the configuration changes of all protein–ligand complexes were assessed in terms of Root Mean Square Deviation (RMSD). The coordinates of the protein backbone and ligand were determined using RMSD analysis to determine how much the ligand location has changed in comparison to the protein. In Fig. 1a, it was observed that the RMSD trajectory of the native proteins 2AX6, 1Z95, and BothMut fluctuated in the first 0-10 ns and 40-60 ns during the simulation and exhibited steady trajectories. Moreover, the RMSD fluctuation analysis in Fig. 1a demonstrates that the MD trajectories of the native proteins were typically stable and within acceptable bounds for the whole studied complex throughout the simulation time. In Fig. 1b, the 2AX6 + GlaMod2 complex (black) and BothMut + GlaMod2 complex (green) showed less fluctuation throughout the simulation and their fluctuating patterns were close to that of the reference complex 2AX6 + FLU (yellow). The complex 2AX6 + GlaMod2 (black) fluctuated initially at 20-40 ns before reaching stability. However, 1Z95 + BIC (blue) and 1Z95 + GlaMod2 (red) fluctuated throughout the simulation. In conclusion, the RMSD fluctuation analysis in Fig. 1b reveals that the MD trajectory of the 2AX6 + GlaMod2 and BothMut + GlaMod2 complex was usually stable and within acceptable bounds during the simulation period throughout the whole studied complex. An additional file shows the other plots of RMSD for native protein and all protein–ligand complexes (See Supplementary Information, Fig. S4). The RMSD plot for native protein and all protein–ligand complexes is shown in Fig. 1a&b.

RMSD along MD trajectories for 100 ns. a RMSD plots of native proteins 2AX6 (black), 1Z95 (red), and BothMut (green); b RMSD plots of complexes 2AX6 + GlaMod2 (black), 1Z95 + GlaMod2 (red), BothMut + GlaMod2 (green),1Z95 + BIC (blue) and 2AX6 + FLU (yellow)
Root Mean Square Fluctuation (RMSF)
The average variance of each amino acid in the protein was calculated using RMSF. Root Mean Square Fluctuation (RMSF) plots were used to show the structure's flexibility for all of the chosen compounds with native receptors at 310 K for 100 ns. The RMSF plot in Fig. 2a revealed that the secondary conformations of the natural proteins 2AX6, 1Z95, and BothMut were stable throughout a 100 ns simulation. During the simulation, the native proteins 2AX6 (black), 1Z95 (red), and BothMut (green) fluctuated below ~ 0.2 nm and were observed to show fluctuations at 2500–3000 atoms. The native protein's RMSF (2AX6, 1Z95, and BothMut) had minimal fluctuation, indicating high stability. The RMSF plot in Fig. 2b indicated that the secondary conformations of the complexes 2AX6 + GlaMod2, 1Z95 + GlaMod2, BothMut + GlaMod2, 1Z95 + BIC and 2AX6 + FLU were stable throughout a 100 ns simulation. It was observed that the complexes 2AX6 + GlaMod2 (black), 1Z95 + GlaMod2 (red), and BothMut + GlaMod2 (green) showed less fluctuation when compared to the reference complexes 1Z95 + BIC (blue) and 2AX6 + FLU (yellow) at 2500–3000 atoms. An additional file shows the RMSF plot for ligand (See Supplementary Information, Fig. S4). The RMSF plot for native protein and all protein–ligand complexes is shown in Fig. 2a&b.

RMSF along MD trajectories for 100 ns. a RMSF plots of native proteins 2AX6 (black), 1Z95 (red), and BothMut (green); b RMSF plots of complexes 2AX6 + GlaMod2 (black), 1Z95 + GlaMod2 (red), BothMut + GlaMod2 (green),1Z95 + BIC (blue) and 2AX6 + FLU (yellow)
Radius of Gyration (Rg)
The compactness of the structures was reflected in the radius of gyration (Rg) plots for all of the chosen compounds with 2AX6, 1Z95, and BothMut receptors at 310 K for 100 ns. The Rg plot in Fig. 3a revealed that in terms of compactness and consistent quantities of Rg, the native proteins 2AX6 (black), 1Z95 (red), and BothMut (green) were found to exhibit extremely similar behavior, suggesting strong stability. The Rg plot in Fig. 3b revealed that in terms of compactness and consistent quantities of Rg, the complexes 2AX6 + GlaMod (black), 1Z95 + GlaMod2 (red), and BothMut + GlaMod2 were found to exhibit similar behavior to their reference complexes 2AX6 + FLU (yellow). However, the complex 1Z95 + BIC (blue) was observed to have more fluctuations over time thereby indicating its instability. The Rg plot for native protein and all protein–ligand complexes is shown in Fig. 3 a&b.

Rg plot along MD trajectories during 100 ns MD Simulation. a Rg plots of 2AX6 protein (black), 1Z95 protein (red), and BothMut protein (green); b Rg plots of complexes 2AX6 + GlaMod2 (black), 1Z95 + GlaMod2 (red), BothMut + GlaMod2 (green), 1Z95 + BIC (blue), and 2AX6 + FLU (yellow)
MM-PBSA
In molecular dynamics simulation, calculating the binding free energy of the lead molecules is always necessary for estimating their binding affinity at different time scales. The binding energy of the selected complexes was investigated using the MM/PBSA process. The binding energy of 2AX6 + GlaMod2 complex (− 85.725 kJ/mol), 1Z95 + GlaMod2 complex (− 67.734 kJ/mol), BothMut + GlaMod2 complex (2935.376 kJ/mol), 1Z95 + BIC reference complex (− 60.766 kJ/mol), 2AX6 + FLU reference complex (− 59.560 kJ/mol), respectively are shown in (Table 4). The van der Waal energy of the reference 1Z95 + BIC (− 108.456 kJ/mol) had a less binding affinity when compared with complexes 2AX6 + GlaMod2 (− 183.681 kJ/mol) and 1Z95 + GlaMod2 (− 118.257 kJ/mol) respectively, showing a strong binding affinity. Electrostatic energy, polar solvation, and SASA energy all had a moderate influence on binding energy in the complexes 2AX6 + GlaMod2, 1Z95 + GlaMod2, and BothMut + GlaMod2. When compared to the reference complex 1Z95 + BIC (− 60.766 kJ/mol) and 2AX6 + FLU (− 59.560 kJ/mol), the binding energy of the complexes 2AX6 + GlaMod2 complex (− 85.725 kJ/mol) and 1Z95 + GlaMod2 complex (− 67.734 kJ/mol) exhibited greater binding affinity. The binding energy demonstrated complex stability, suggesting that complexes (2AX6 + GlaMod2 and BothMut + GlaMod2) might be considered potential inhibitors of T877A and W741L mutations in Prostate Cancer.
The ligands’ average free binding energy (∆G) was calculated using the MM/PBSA technique, and graphs were created using QtGrace software. The ∆G-binding energy of the ligands is shown in Fig. 4

The average free binding energy of the ligands in complexes (2AX6 + GLA, 2AX6 + GLAMOD2, 1Z95 + GLA, 1Z95 + GLAMOD2, BothMut + GLA and 1Z95 + BIC
QSAR Prediction
Hansch and Fujita developed the QSAR approach based on the work of Hammett and Taft [54]. The Quantitative Structure–Activity Relationship Study (QSAR) models are regression or classification models that predict the activities of novel chemical compounds based on their physicochemical features. Generally, generally, QSAR is a regression model that connects the potency of the response variable (Y) like the biological activity of the molecule to a collection of 'predictor' variables (X). The QSAR illustrates the association between molecular descriptors (chemical structures) that reflect the distinctive physicochemical qualities of compound sets of interest and their biological activity. To predict the behavior of the GlaMod2 compound with mutant ARs in prostate cancer, the compound was submitted to QSAR analysis. The goal of this study was to investigate if the GlaMod2 compound might be utilized as an inhibitor against mutant ARs in prostate cancer by looking at its inhibitory activity. To assess the efficacy of various drugs, the Quantitative Structure–Activity Relationship Study is commonly employed [55]. In QSAR analysis, the drug targets were correlated against seven molecular descriptors: Molecular weight, LogP, Polarizability, Refractivity, Molecular Surface area, Polar Surface Area, and Hydrophilic-Lipophilic Balance (HLB). The statistical analysis of the results of QSAR from (Table S3 in the supplementary) built a QSAR model that showed the Sum of squares due to Regression (SSR) = 26.38, Sum of squared errors (SSE) = 8.31, a total sum of squares (SST) = 34.69, R2 value of 76.04 percent and an adjusted R2 value of 65.55 percent showing very less difference between R2 and adjusted R2. The Rsq/R2 value is high predicting good QSAR. Greater Rsq/R2 indicates that the equation fits the data better. The F-statistics value was found to be 7.25 which was higher than the F-critical value of 2.44. The percentage contribution of each descriptor to the activity obtained was: Molecular Weight (5.26%), AlogP (37.54%), Polarizability (5.11%), Refractivity (33.80%), Molecular Surface Area (5.33%), Polar Surface Area (11.64%), HLB (23.29%) respectively. EasyQSAR software was used to create the correlation and regression. EasyQSAR software was utilized in this study to perform QSAR analysis on 30 known prostate cancer drug targets. The ChEMBL online database was used to determine the IC50 values of 30 known prostate cancer drug targets. An additional file depicts the descriptors and function of QSAR of 30 known drug targets of prostate cancer, which is divided into 24 training and 6 test sets in tabular form (See Online resource, Table S3). The Multiple Regression Plot generated for the QSAR model is represented in supplementary (Supplementary Information, Fig S4). The selected ligands’ behavior was then predicted using a QSAR-generated regression equation which is as follows:
By considering this equation the activity of the potential drug candidate GlaMod2 was found and was converted by taking the antilogarithm of the inverse of predicted activity to find the potency. The Descriptors and predicted IC50 values of the GlaMod2 compound have been represented in Table 5.
Table 5 shows the descriptors and projected IC50 values of the GlaMod2 compound produced by QSAR analysis. The lower the IC50 value, the more potent they are. The IC50 values of GlaMod2 were found to be (20.89 nM).
From the Molecular Docking, Drug Likeliness capacity, ADMET test, Molecular Dynamics Simulations, and QSAR study the compound GlaMod2 was observed as a potential drug candidate having more potency as an inhibitor of mutated AR of Prostate Cancer. However, to further strengthen the findings, the potential ligand can be synthesized to perform in-vitro studies for further evaluation.
Conclusion
This research has been carried out due to the fact that cancer drug resistance remains a severe issue, causing the most deterioration and ranking among the top causes of cancer mortality. In prostate cancer, drug resistance develops when tumor cells become less sensitive and resist chemotherapy and androgen deprivation treatment (ADT). It is one of the most challenging difficulties in the treatment of individuals with advanced prostate cancer (PCa). To combat drug resistance and improve clinical outcomes, new and effective drugs must be developed. Designing novel drugs has been recognized as a demanding, costly, time-consuming, and difficult process. In such instances, computer-aided drug discovery (CADD) has emerged as a potent and promising technique for the pharmaceutical industry to design drugs faster, cheaper, and more effectively. The fast development of computational techniques for drug discovery has also yielded valuable insights into cancer therapy.
The current work focussed on the alteration of plant-derived flavonoids, which may function as inhibitors against T877A and W741L point mutations using computational tools. In this study, the compound GlaMod2 was found to be effective against T877A and W741L mutations in prostate cancer patients. This phytocompound may be beneficial in the development of prostate cancer inhibitors for patients with mutant androgen receptors. Computational techniques such as Molecular Docking, Molecular Dynamics Simulation, MM-PBSA, and QSAR analysis were often employed in this study. In contrast, bioassays employing prostate cancer cell lines and animal models could be performed to further validate this lead compound. In-depth in-vivo research can therefore help in understanding the intended drug candidate’s behavior in the metabolic system and determining the in-vivo toxicity impact.
Availability of Data and Materials
All data generated or analyzed during this study are included in this manuscript and its online resource file.
References
Batiha GES, Beshbishy AM, El-Mleeh A, Abdel-Daim MM, Devkota HP. Traditional uses bioactive chemical constituents, and pharmacological and toxicological activities of Glycyrrhiza glabra L Fabaceae. Biomolecules. 2020;10(3):352. https://doi.org/10.3390/biom10030352.
Kreyling WG, Semmler M, Erbe F, Mayer P, Takenaka S, Schulz H, et al. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J Toxicol Environ Health A. 2002;65(20):1513–30. https://doi.org/10.1080/00984100290071649.
El-Saber Batiha G, Magdy Beshbishy A, El-Mleeh A, Abdel-Daim MM, Prasad Devkota H. Traditional uses, bioactive chemical constituents, and pharmacological and toxicological activities of Glycyrrhiza glabra L. (Fabaceae). Biomolecules. 2020;10(3):352. https://doi.org/10.3390/biom10030352.
Wang L, Yang R, Yuan B, Liu Y, Liu C. The antiviral and antimicrobial activities of licorice, a widely-used Chinese herb. Acta Pharm Sin B. 2005;5(4):310–5. https://doi.org/10.1016/j.apsb.2015.05.005.
Varshney IP, Jain DC, Srivastava HC. Study of Saponins from Glycyrrhiza glabra Root. Int J Crude Drug Res. 1983;21(4):169–72. https://doi.org/10.3109/13880208309070637.
Shinde DB, Koratkar SS, Sharma N, Shitole AA. Antioxidant activity and antiproliferative action of methanolic extract of liquorice (Glycyrrhiza glabra) in HEPG2 cell line. Int J Pharm Pharm Sci. 2016. https://doi.org/10.22159/ijpps.2016v8i9.11954
Govind P, Sharma M. Ethnomedicinal plants for prevention and treatment of tumors. Int J Green Pharm. 2009;3(1):2. https://doi.org/10.4103/0973-8258.49367.
Pilcher H. Liquorice may tackle SARS. Nature. 2003. https://doi.org/10.1038/news030609-16.
Young S-M, Bansal P, Vella ET, Finelli A, Levitt C, Loblaw A. Systematic review of clinical features of suspected prostate cancer in primary care. Can Fam Physician. 2015;61(1):e26-35.
Kaler J, Hussain A, Haque A, Naveed H, Patel SA. Comprehensive review of pharmaceutical and surgical interventions of prostate cancer. Cureus. 2020;12(11):e11617. https://doi.org/10.7759/cureus.11617.
Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harbor Perspectives in medicine 2016;6(3), a026104. https://doi.org/10.1101/cshperspect.a026104.
Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer. 2011;2(4):466–74. https://doi.org/10.1177/1947601911408889.
Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinogenesis. 2011;10:20. https://doi.org/10.4103/1477-3163.83937.
Rathkopf DE, Smith MR, Ryan CJ, Berry WR, Shore ND, Liu G, Higano CS, Alumkal JJ, Hauke R, Tutrone RF, Saleh M, Chow Maneval E, Thomas S, Ricci DS, Yu MK, de Boer CJ, Trinh A, Kheoh T, Bandekar R, Scher HI, Antonarakis ES. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Annals Oncol Off J European Soci Med Oncol. 2017;28(9):2264–71. https://doi.org/10.1093/annonc/mdx283.
Sakkiah S, Kusko R, Pan B, Guo W, Ge W, Tong W, Hong H. Structural changes due to antagonist binding in ligand binding pocket of androgen receptor elucidated through molecular dynamics simulations. Front Pharmacol. 2018. https://doi.org/10.3389/fphar.2018.00492.
Nadiminty N, Gao AC. Mechanisms of persistent activation of the androgen receptor in CRPC: Recent advances and future perspectives. World J Urol. 2012; 30, 287–295.
Terakawa T, Miyake H, Kumano M, Sakai I, Fujisawa M. The antiandrogen bicalutamide activates the androgen receptor (AR) with a mutation in codon 741 through the mitogen activated protein kinase (MARK) pathway in human prostate cancer PC3 cells. Oncol Rep. 2010;24(5):1395–9. https://doi.org/10.3892/or_00000998.
Singh AN, Baruah MM, Sharma N. Structure-Based docking studies towards exploring the potential anti-androgen activity of selected phytochemicals against Prostate Cancer. Sci Rep. 1955; 7(1). https://doi.org/10.1038/s41598-017-02023-5.
Tian S, Wang J, Li Y, Li D, Xu L, Hou T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv Drug Deliv Rev. 2015;86:2–10. https://doi.org/10.1016/j.addr.2015.01.009.
Salsbury FR. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr Opin Pharmacol. 2010;10(6):738–44. https://doi.org/10.1016/j.coph.2010.09.016.
Hansch C, Verma RP. Overcoming tumor drug resistance with C2-modified 10-deacetyl-7-propionyl cephalomannines: a QSAR study. Mol Pharm. 2009;6(3):849–60. https://doi.org/10.1021/mp800138w.
Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J Biol Chem. 2005;280(45):37747–54. https://doi.org/10.1074/jbc.M507464200.
Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci. 2005;102(17):6201–6. https://doi.org/10.1073/pnas.0500381102.
Matias PM. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem. 2000;275(34):26164–71. https://doi.org/10.1074/jbc.m004571200.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12. https://doi.org/10.1002/jcc.20084.
National Center for Biotechnology Information. PubChem Compound Summary for CID 124049, Glabranin. Retrieved March 17, 2023 from https://pubchem.ncbi.nlm.nih.gov/compound/Glabranin.
National Center for Biotechnology Information. PubChem Compound Summary for CID 91649, Hydroxyflutamide. Retrieved March 17, 2023 from https://pubchem.ncbi.nlm.nih.gov/compound/Hydroxyflutamide.
National Center for Biotechnology Information. PubChem Compound Summary for CID 56069, (R)-Bicalutamide. Retrieved March 17, 2023 from https://pubchem.ncbi.nlm.nih.gov/compound/R_-Bicalutamide.
National Center for Biotechnology Information. PubChem Compound Summary for CID 2375, Bicalutamide. Retrieved March 17, 2023 from https://pubchem.ncbi.nlm.nih.gov/compound/Bicalutamide.
National Center for Biotechnology Information. PubChem Compound Summary for CID 3397, Flutamide. Retrieved March 17, 2023 from https://pubchem.ncbi.nlm.nih.gov/compound/Flutamide.
ChemAxon - Software Solutions and Services for Chemistry & Biology. Chemaxon.com. 2021. https://chemaxon.com/. Accessed 15 Dec 2021.
Yang Z, Lasker K, Schneidman-Duhovny D, Webb B, Huang CC, Pettersen EF, et al. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol. 2012;179(3):269–78. https://doi.org/10.1016/j.jsb.2011.09.006.
Biswas SS, Browne RB, Borah VV, Roy JD. In Silico approach for phytocompound-based drug designing to fight efflux pump-mediated multidrug-resistant mycobacterium tuberculosis. Appl Biochem Biotechnol. 2021;193(6):1757–79. https://doi.org/10.1007/s12010-021-03557-1.
Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. In: Chemical biology, 2015. pp. 243–250. Humana Press, New York, NY.
Ramezani A, Zakeri A, Mard-Soltani M, Mohammadian A, Hashemi ZS, Mohammadpour H, Jahangiri A, Khalili S, Rasaee MJ. Structure based screening for inhibitory therapeutics of CTLA-4 unveiled new insights about biology of ACTH. Int J Peptide Res Therapeut. 2019. https://doi.org/10.1007/s10989-019-09891-7.
Souza PF, Lopes FE, Amaral JL, Freitas CD, Oliveira JT. A molecular docking study revealed that synthetic peptides induced conformational changes in the structure of SARS-CoV-2 spike glycoprotein, disrupting the interaction with human ACE2 receptor. Int J Biol Macromol. 2020;164:66–76.
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–86.
Molinspiration Cheminformatics. Choice (Middletown). 2006. 43(11):43-6538-43-6538. https://www.molinspiration.com/. Accessed 15 Dec 2021.
Drug Likeness Tool (DruLiTo 1). Gov. in. 2021. http://www.niper.gov.in/pi_dev_tools/ DruLiToWeb/DruLiTo_index.html. Accessed 15 Dec 2021.
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–86. https://doi.org/10.1021/ci200227u.
Bekker H, Berendsen HJC, Dijkstra EJ, Achterop S, Vondrumen R, Vanderspoel D, Sijbers A, Keegstra H, Renardus MKR. Gromacs-a parallel computer for molecular-dynamics simulations. In: 4th International Conference on Computational Physics (PC 92). 1993; pp. 252–256. World Scientific Publishing.
Kumari R, Kumar R. Open-source drug discovery consortium, Lynn A. g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model. 2014;54(7):1951–62. https://doi.org/10.1021/ci500020m.
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, et al. 3rd, calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33(12):889–97. https://doi.org/10.1021/ar000033j.
Davies M, Nowotka M, Papadatos G, Dedman N, Gaulton A, Atkinson F, et al. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015;43(W1):W612–20. https://doi.org/10.1093/nar/gkv352.
Gerçek Z, Ceyhan D, Erçağ E. Synthesis and molecular docking study of novel COVID-19 inhibitors. Turk J Chem. 2021;45(3):704. https://doi.org/10.3906/kim-2012-55.
Kuntal. EasyQSAR: A beginners tool for QSAR in Drug Designing (Free software for drug designing and QSAR). Blogspot.com. 2021. http://easyqsar.blogspot.com/2009/09/easyqsar-beginners-tool-for-qsar-in.html. Accessed 16 Dec 2021.
Banu BH, Rajitha G, Bharathi K. An approach to computer aided drug design of some bioactive cinnamoyl hydrazones, in silico and docking studies as possible COX-2 selective inhibitors. J Biotechnol Biomater. 2016. https://doi.org/10.4172/2155-952x.1000240.
Leeson PD, Davis AM. Time-related differences in the physical property profiles of oral drugs. J Med Chem. 2004;47(25):6338–48. https://doi.org/10.1021/jm049717d.
Muchmore SW, Edmunds JJ, Stewart KD, Hajduk PJ. Cheminformatic tools for medicinal chemists. J Med Chem. 2010;53:4830–41.
Selvam P, De S, Paira P, Kumar SA, Moorthy A, Ghosh A, Jenifer SK. In vitro studies on the selective cytotoxic effect of luminescent Ru (II)-p-cymene complexes of imidazo-pyridine and imidazo quinoline ligands. Dalton Trans. 2022;51(45):17263–76.
De S, Kumar RS, Gauthaman A, Kumar SA, Paira P, Moorthy A, Banerjee S. Luminescent ruthenium(II)-para-cymene complexes of aryl substituted imidazo-1,10-phenanthroline as anticancer agents and the effect of remote substituents on cytotoxic activities. Inorganica Chimica Acta. 2021;515:120066. https://doi.org/10.1016/j.ica.2020.120066.
Pandey B, Grover A, Sharma P. Molecular dynamics simulations revealed structural differences among WRKY domain-DNA interaction in barley (Hordeum vulgare). BMC Genom. 2018. https://doi.org/10.1186/s12864-018-4506-3.
Browne RB, Goswami N, Borah P, Roy JD. Computational approaches for evaluation of isobavachin as potential inhibitor against t877a and w741l mutations in prostate cancer. J Biomol Struct Dyn. 2022; https://doi.org/10.1080/07391102.2022.2032353.
Fujita T, Iwasa J, Hansch C. A new substituent constant, π, derived from partition coefficients. J Am Chem Soc. 1964;86(23):5175–80.
Abdel-Ilah L, Veljović E, Gurbeta L, Badnjević A. Applications of QSAR study in drug design. Int J Eng Res Technol (IJERT). 2017; 6(06).
Fujita K, Nonomura N. Role of androgen receptor in prostate cancer: a review. World J Mens Health. 2019;37(3):288–95. https://doi.org/10.5534/wjmh.180040.
Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20(13):3001–15. https://doi.org/10.1200/jco.2002.10.018.
Acknowledgements
We acknowledge the Bioinformatics Infrastructure Facility, C.V.Sc., Khanapara, Guwahati, sponsored by the Department of Biotechnology, Ministry of Science and Technology, Govt. of India for providing facilities to conduct molecular dynamics studies in a high-performance Dell Precision Tesla 5600 workstation with 12 CPUs and 1 NVIDIA Tesla C2075 graphics processing unit (GPU). We would also like to thank the Vice Chancellor, Assam Don Bosco University for providing the research facilities. The authors appreciate the continuous encouragement and advice from the Director, School of Life Science, Assam Don Bosco University.
Funding
The authors declare that no funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript. R. B. B. has designed and compiled the Manuscript. J. D. R. has conceived and supervised the research. N. G. has guided the computational work and also in performing the Molecular Dynamics Simulation studies. P. B. who is the Coordinator, BIF, and Head of the Department of Animal Biotechnology, College of Veterinary Science has guided and allowed to perform molecular dynamics simulation studies in a high-performance workstation in his lab. All these above-mentioned authors contributed equally to the successful completion of the research work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent to Publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Browne, R.B., Goswami, N., Borah, P. et al. Study of Glabranin as an Inhibitor Against Prostate Cancer: Molecular Docking, Molecular Dynamics Simulation, MM-PBSA Calculation and QSAR Prediction. Ind J Clin Biochem 39, 331–343 (2024). https://doi.org/10.1007/s12291-023-01134-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12291-023-01134-3