
Abstract
Background
Splicing factor 3b subunit 1 (SF3B1) was frequently reported to be significantly mutated in breast cancer. However, the status of SF3B1 expression, its function and molecular consequence in breast cancer remained unreported.
Methods
Immunohistochemistry was used to assess SF3B1expression in 110 breast cancer samples. SF3B1 knock‑down in ZR-75-30 and MDA-MB-231 cells was performed by shRNA transfection. The expression of SF3B1 in cells was detected by quantitative real‑time PCR and western blot. Cell proliferation ability was determined by MTT and colony formation assay. Migration and invasion were determined by transwell assay. Flow cytometry was performed to investigate cell cycle and apoptosis. RNA-sequencing was performed to examine differentially expressed genes and affected alternative splicing events.
Results
SF3B1 is overexpressed in breast cancer tissues compared with normal tissues. Overexpression of SF3B1 is associated with lymph node metastasis. SF3B1 knockdown in MDA-MB-231 and ZR-75-30 breast cancer cells significantly induced the suppression of proliferation, migration, invasion and also enhancement of apoptosis. RNA-sequencing data revealed that 860 genes were significantly up-regulated and 776 genes were significantly down-regulated upon SF3B1 knockdown. Differentially expressed genes enriched in the signaling pathways including Ras signaling pathway; cytokine receptor interaction; tight junction; MAPK signaling pathway, Glycine, serine and threonine metabolism. Alternative splicing analysis revealed that exon skipping (SKIP) and cassette exons (MSKIP) were the most common molecular effect upon SF3B1 knockdown.
Conclusions
Our study suggests that SF3B1 may be an important molecular target for breast cancer treatment and provides a new clue for clinical treatment of breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Abnormal splicing is one of the hallmarks of cancer-associated phenotypes [1]. It is well known that alternative splicing (AS) is controlled by spliceosome which is comprised of small nuclear RNAs (snRNA) and various proteins [2]. Mutation and expression level change of splice factor genes can result in loss of exons, intron retention and misidentification of splice sites, thereby affecting post-transcriptional processing and expression of most genes involved in cell cycle control, angiogenesis, apoptosis, metastasis and other processes. Many cancers have lots of alternative splicing events that do not exist in normal samples. Therefore, targeting splicing deregulation could represent one of novel ways for cancer therapy.
The utilization of next-generation sequencing has revealed the significantly somatic mutations of splicing factors in multiple cancers. Among them, the SF3B1 mutations occurred in breast, endometrial cancers, uveal melanomas and several types of hematological malignancies [3,4,5,6,7]. The independent next-generation sequencing of 892 and 510 breast cancer samples revealed significant and frequent mutation of SF3B1, especially in patients with estrogen receptor (ER)-positive subtypes [8, 9]. Of these mutations, missense mutation is dominated and most of missense mutations locate in some repetitive sites such as K700E and Q534P hotspot. SF3B1 gene is located in chromosome 2q33.1 and encodes the subunit 1 of splicing factor 3b, a core part of the U2 small nuclear ribonucleoprotein complex. SF3B1 is essential for recognizing and binding the branch point sequences close to 3′ splice sites and plays an important role in the accurate excision of introns from pre-mRNA to form mature mRNA [10]. SF3B1 mutation may be a driving event in tumorigenesis, not only causing splicing abnormalities in transcriptional coupling, but also affecting genomic instability and differentiation of stem cells [3, 11, 12].
Breast cancer is known to be a heterogeneous disease driven by a large repertoire of molecular abnormalities, which contribute to its diverse clinical behavior. Despite the success of targeted therapy approaches for breast cancer management, drug resistance remains a very serious problem and the molecular mechanism of breast cancer still needs to be clarified. Although SF3B1 mutations have been frequently reported in breast cancer, the status of SF3B1 expression, its function and molecular consequence in breast cancer remained unreported. In this study, our aims were to assess the expression level of SF3B1 in breast cancer and the relationship with clinical and pathological features. Moreover, we explored the biological effects and downstream molecular events after SF3B1 knockdown in breast cancer cells. Our results suggest the therapeutic potential of SF3B-targeting compounds against breast cancer.
Materials and methods
Samples and clinical data
Cancer tissue samples from 110 patients with diagnosed breast cancer were obtained from the first affiliated hospital of Shanxi medical university according to the guidelines of the local ethics committees, forty of which had matched non-tumor control tissues. The molecular subtypes covered Luminal A (38), Luminal B (38), HER2-enriched (14) and Triple-negative/basal-like breast cancer (20). No patients had received radiotherapy or chemotherapy before the surgery.
Immunohistochemistry
Immunohistochemistry of paraffin-embedded formalin-fixed tissue samples was performed as previously described [13]. Specimens were sliced into 4 μm-thick sections. The sections were deparaffinized and rehydrated using gradient xylene and alcohol. Then the samples were incubated with 3% H2O2 for 10–15 min and rinsed with distilled water three times. After antigen retrieval in sodium citrate buffer (pH 6.0) for 2 min and blocking in goat serum, sections were incubated with specific anti-SF3B1 antibody (Abcam) at a 1:200 dilution overnight at 4 °C. After washing with PBS, the sections were incubated with second antibody (Maixin) at 37 °C for 20 min, followed by detection using the DAB detection kit (Maixin). The expression levels of SF3B1 were analyzed using fully automatic digital pathological scanning apparatus (Aperio, Vista, CA, USA). Histochemistry score (H-score), also known as immunoreactivescore (IRS), was automatically generated by Aperio Nuclear v.9 software and calculated by assessment of both the staining intensity and the percentage of positive cells.
Cell culture
MDA-MB-231 and ZR-75-30 breast cancer cell lines were cultured in L-15 and RPMI-1640 medium supplemented with 10% fetal bovine serum and antibiotics composed of 1% penicillin/streptomycin. The cultures were kept at 37 °C in a humidified atmosphere of 95% air and 5% CO2.
SF3B1 knockdown in breast cancer cell lines
For knockdown of endogenous SF3B1, two lentivirus shRNA sequences were used: 5′-ccggGAGC ACAGACCTCCAAAGATTctcgagAATCTTTGGAGGTCTGTGCTCtttttg-3′ (named as SF3B1-sh1) and 5′-ccggTGCAACTGAACTTGAAGATGActcgagTCATCTTCAAGTTCAGTTGCAtttttg-3′ (named as SF3B1-sh2). These shRNA sequences were cloned into GV248 vector (hU6-MCS- Ubiquitin-EGFP -IRES-puromycin) and co-transfected into 293 T cells along with packaging plasmids by using Lipofectamine 2000 reagent (Invitrogen). To lentivirus transduction, breast cancer cells were seeded at 30–40% confluence and infected with appropriate titer of virus and incubated overnight.
Western Blot
The protein was isolated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred to nitrocellulose filter membranes and the membranes were incubated with 5% skimmed milk at room temperature for 1 h. Then the membranes were incubated with primary antibody (Abcam) at 4 °C overnight. The blot was detected with horseradish peroxidase labeled secondary antibody (Sigma), and chemiluminescence was detected with a LAS4000 device (Fuji).
Real-time quantitative PCR (qPCR)
Total RNA was extracted from cell lines with Trizol reagent according to the manufacturer’s instruction. Then total RNA was reverse transcribed into cDNA by TaqMan RT Kit according to manufacturer’s instruction. cDNA was amplified by PCR with 20 μl reaction system using SYBR-Green PCR Master Mix according to manufacturer’s instruction. Primers for SF3B1 (F 5′-GAGAAATTCAAGGCAAG AAGG-3′; R 5′-GACCAAGCAAACTCGTAGATG-3′); GAPDH (F 5′-TGACTT CA ACAGCGACA CCCA-3′; R 5′-CACCCTGTTGCTGTAGCCAAA-3′) were used. All RT-qPCR reactions were completed in triplicate.
MTT assay
Cells were seeded into a 96‑well plate at a density of 4 × 103 cells/well and incubated in normal conditions. At the specified time, 20 μl MTT solution (5 mg/ml) was added to each well and cells were incubated for 4 h until crystals were formed. The medium was then removed and replaced with 200 μl DMSO. After shaking for fifteen minutes until crystals were dissolved, the optical density was determined by Microplate Reader (Bio-Rad) at 490 nm.
Colony formation, migration and invasion assay
500–800 cells were seeded per well in 6-well plates containing complete medium and cultured in normal conditions. On day 14, cells were fixed with 4% polyformaldehyde for 15 min and stained with 1% crystal violet for twenty minutes. Then the samples were slowly washed with running water. Transwell assay was used to evaluate the migration and invasion ability of breast cancer. The bottom membrane of transwell chambers with or without Matrigel Basement Membrane Matrix (BD) was used for invasion and migration detection. 2–3 × 104 cells in serum-free medium were seeded into the upper chambers and the lower chambers were added with serum-containing medium. After 48 h of culture, the cells through the membrane were fixed with 4% methanal for 20 min and stained with 1% crystal violet for 20 min and counted under a microscope.
Apoptosis and cell cycle assay
Collected cell precipitation in 6-well plates was washed using 4 °C pre-cooled D-Hanks and 1 × binding buffer. Cell pellet was re-suspended in 200 μl of 1 × binding buffer and stained using 10 μl of Annexin V-APC in the dark for 10–15 min at room temperature and detected by flow cytometry. The cells in 6 cm dish were collected and were centrifuged. The supernatant was discarded, followed by the addition of 4 °C pre-cooled D-Hanks. After centrifugation, precooled 70% ethanol was used to fix cells overnight at 4 °C. After being washed, RNase-containing PI solution was added and incubated for 30 min in the dark. The analysis was performed with a flow cytometer (BD Biosciences, San Diego, CA, USA).
RNA sequencing
RNA sequencing was performed in Shanghai Genechem Co., LTD. After the total RNA was qualified, the mRNA was enriched using magnetic beads with Oligo (dT). The extracted mRNA was randomly fragmented and used as a template to synthesize cDNA with random primers, followed by double-stranded cDNA synthesis with buffer, dNTPs, RNaseH and DNA polymerase. The products were purified by AMPure XP beads. Then, the T4 DNA polymerase and Klenow DNA polymerase were used to repair the cohesive end of DNA to blunt end, followed by adding base A and adaptor to 3′ end. Finally, PCR amplification was performed to obtain a final sequencing library. After the library was qualified, Illumina Hiseq4000 was used to sequence and the sequencing read length was double-ended 2 × 150 bp (PE150).
Statistical analysis
Rank sum and Chi square (\(\chi {2}\)) tests were carried out to compare differences of SF3B1 expression with clinicopathological parameters. Measurement data were expressed as mean±SD. Analyses were performed using SPSS.20 statistical software. Student’s t-test was used to compare differences between two groups. One- or two-way ANOVA was used to compare differences among three or more groups. A p value less than 0.05 was considered statistically significant.
Results
Relationships between SF3B1 expression and clinicopathological characteristics
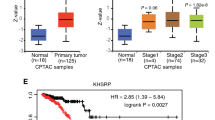
First, SF3B1 protein levels were measured in 110 breast cancer tissues. The results of immunohistochemistry showed that SF3B1 was expressed in the nucleus of breast cancer cells (Fig. 1a). The H-Score of SF3B1 protein ranged from 1.38278 to 196.3014 in breast cancer tissues, and the median was 162.7205. The SF3B1 protein expression in cancer tissues showed significantly higher than that of non-tumor tissues (p < 0.01) (Fig. 1a–b). Moreover, according to the ROC curve analysis (Fig. 1c), the optimal cut-off value of 71.2758 of SF3B1 expression level was selected with higher sensitivity and specificity to divide all breast cancer cases into two groups: SF3B1-low (H-score < 71.2758) and SF3B1-high (H-score ≥ 71.2758). Our analysis showed that SF3B1 expression level was associated with lymph node metastasis (p < 0.05) and no obvious correlations were seen in patients of different age, clinical stage, tumor size, status of estrogen, progesterone, HER-2, Ki-67 (Table 1). In addition, TCGA data analysis showed that expression level of SF3B1 in luminal subtype which represents ER-positive breast cancer is significantly higher than that of HER2-positive breast cancer (p < 0.01) (Fig. 1d).

SF3B1 was up-regulated in breast cancer tissues compared to that of adjacent non-tumor tissues. a Representative immunohistochemistry images of SF3B1 expression. Scale bars represent 50 μm. b Analysis of SF3B1 expression level based on immunohistochemistry data (Rank sum test, p < 0.0001). The Y axis represents the H-score automatically generated by Aperio Nuclear v.9 software based on staining intensity and the positive percentage. c ROC curve analyses of SF3B1 expression in breast cancer (AUC = 0.787, p < 0.0001). d SF3B1 expression in breast cancer subtype from TCGA database
Effects of SF3B1 knockdown on the cell proliferation and colony formation
To further define the biological function of SF3B1 on breast cancer, we measured expression level of SF3B1 in six breast cancer cell lines by RT-qPCR and found different expression level in different cell lines (Fig. 2a). In order to see if there was difference in the role of SF3B1 in estrogen-positive and estrogen-negative breast cancer cells, MDA-MB-231 and ZR-75-30 cell lines with high expression of endogenous SF3B1 were used for knockdown experiment.

Knockdown of SF3B1 inhibits breast cancer cell proliferation and colony formation in vitro. a The mRNA expression pattern of SF3B1 in 6 of breast cancer cell lines detected by RT-PCR. b Knockdown efficiency of SF3B1 were tested by qPCR. c Knockdown efficiency of SF3B1 were tested by western blot. d SF3B1 knockdown inhibited the proliferation of ZR-75-30 and MDA-MB-231 cells (p < 0.01). e The number of colony formation was decreased in SF3B1 knockdown group compared to the control (p < 0.01)
As showed in Fig. 2b, c, the knockdown efficiency was confirmed by RT-qPCR and western blot in ZR-75-30 and MDA-MB-231 cell lines. Then, MTT assay was used to illustrate the relationship between knockdown of SF3B1 and cell proliferation. Absorbance value was measured at 24, 48, 72, 96 and 120 hr. The absorbance value of SF3B1-sh1 and sh2 in ZR-75-30 and MDA-MB-231 cells significantly decreased compared with corresponding negative control group at 72, 96 and 120 hr (p < 0.05) (Fig. 2d). In addition, colony formation efficiency was also decreased in both shRNA‑treated ZR-75-30 and MDA-MB-231 cell lines as shown by the colony formation assay (p < 0.01) (Fig. 2e).
Effects of SF3B1 knockdown on the cell cycle and apoptosis
Flow cytometry was used to investigate the effect of SF3B1 knockdown on cell cycle distribution in ZR-75-30 and MDA-MB-231 cells. The affected cell cycle progression is different in these two cells. For ZR-75-30 cell line, no significant difference was seen between negative control group and SF3B1 knockdown group (Fig. 3a). For MDA-MB-231 cell line, DNA ploidy detection indicated that the rates of G1 and S phase cells were significantly decreased in the SF3B1 knockdown group compared with the negative control group (p < 0.05) (Fig. 3a). However, G2/M phase rates were significantly increased in the SF3B1 knockdown group compared with the negative control group (p < 0.01) (Fig. 3a). The results indicated that SF3B1 knockdown inhibited DNA replication and played an important role in G2/M block. The different results of cell cycle progression upon SF3B1 knockdown in ZR-75-30 and MDA-MB-231 cells may be attributed to the genetic background, of which ZR-75-30 represents luminal B breast cancer cell line and MDA-MB-231 represents basal-like breast cancer cell line.

SF3B1 knockdown affects cell cycle and enhances cell apoptosis in breast cancer. a The cell cycle was investigated by flow cytometry in ZR-75-30 (top) and MDA-MB-231 cells (bottom). b Cell apoptosis of ZR-75-30 (top) and MDA-MB-231 (bottom) were suggested by flow cytometry (p < 0.01)
Next, the effect of SF3B1 knockdown on cell apoptosis was investigated. The Annexin V-APC assay showed that apoptosis rate of SF3B1 knockdown group in ZR-75-30 cells (9.75% ± 0.27% or 9.95% ± 0.47%) significantly increased compared with those of the negative control groups (4.30% ± 0.22%) (p < 0.01) (Fig. 3b). The pro-apoptotic effect upon SF3B1 knockdown is more obvious in MDA-MB-231 cells, in which knockdown group showed significantly increased apoptosis rate compared to negative control groups (22.90 ± 0.79% or 24.90 ± 0.89% vs 1.43 ± 0.21%) (p < 0.001) (Fig. 3b).
Effects of SF3B1 knockdown on the cell migration and invasion
Next, we detected the influence of SF3B1 knockdown on cell invasion in breast cancer using transwell chamber coated with matrigel. As showed in Fig. 4a, cell motility potential in SF3B1 knockdown group cells was significantly decreased compared with negative control group. For ZR-75-30 cells, the relative invasion rate of SF3B1 knockdown group cells was 48.00 ± 4.91 or 40.1 ± 3.92 as compared to 155.10 ± 2.31 of negative control cells (p < 0.01). For MDA-MB-231 cells, the relative invasion rate was more significantly reduced in SF3B1 knockdown cells as compared to negative control group (23.21 ± 0.17 or 32.00 ± 1.15 vs. 170 ± 12.45, p < 0.01) (Fig. 4a).

SF3B1 knockdown inhibits migration and invasion of breast cancer cells. a SF3B1 knockdown markedly inhibited ZR-75-30 (top) and MDA-MB-231 (bottom) cell invasion (p < 0.01). b The migration ability was inhibited by SF3B1 knockdown in ZR-75-30 (top) and MDA-MB-231 (bottom) cells (p < 0.01)
Transwell chamber assay was also used to detect migration ability. In both cell lines, the number of cell permeating in SF3B1 knockdown group cells dropped significantly from control group cells (107.00 ± 0.85 or 99.10 ± 2.88 vs. 153.00 ± 6.37 in ZR-75-30 cells; 3.01 ± 0.26 or 2.13 ± 0.16 vs. 110.00 ± 1.44 in MDA-MB-231 cells) (Fig. 4b).
Downstream pathway and affected alternative splicing events observed following SF3B1 knockdown
In order to examine the effects of SF3B1 knockdown on the whole transcriptome in breast cancer, we extracted the RNA of SF3B1 knockdown and negative control MDA-MB-231 cells and conducted RNA sequencing. Three independent biological replicates were conducted. An average of 52 million paired-end 150-bp sequencing reads per sample were acquired. Differential expression analysis using a significant difference threshold of twofold change revealed that 860 genes were significantly up-regulated and 776 genes were significantly down-regulated in SF3B1 knockdown group compared with negative control (Fig. 5a). The heatmap shows the top 100 differentially expressed genes and clustering of the three replicates according to expression change (Fig. 5b). The pathway enrichment analysis demonstrated that the differentially expressed genes were enriched in the signaling pathways including Ras signaling pathway; cytokine-cytokine receptor interaction; tight junction; MAPK signaling pathway, Glycine, serine and threonine metabolism and so on, indicating that SF3B1 may contribute to breast cancer tumorigenesis via these pathways (p < 0.05) (Fig. 5c). Gene ontology analysis revealed that the differentially expressed genes (p < 0.05) were enriched in biologic processes including regulation of transcription, DNA template or from RNA polymerase II promotor; transport; apoptotic process; cell cycle; cell differentiation and so on (Fig. 5d).

Differentially expressed gene, key cancer pathway and biological process components altered in SF3B1 knockdown cells compared to corresponding control cells analyzed via RNA-sequencing approach. a Volcano plot displaying differentially expressed genes. Significant differences are defined as p value < 0.05 and log2 fold change > 1. Red represents upregulation and blue represents downregulation. b Heatmap of top 100 differentially expressed genes. Each row is a gene and each column is a replicate. c The pathway-enrichment analysis of differentially expressed genes. Gene number and p-value are shown on the right d GO analysis of annotation entries of significantly differentially expressed genes. Gene Ontology includes three ontologies: the biological process (blue), cellular component (green), and molecular function (orange)
Moreover, we applied ASprofile software to RNA-sequencing data to investigate the global effect of SF3B1 knockdown on splicing. We classified the alternative splicing events into seven categories and analyzed the frequency of the splicing pattern changes upon SF3B1 knockdown (Fig. 6a). Alternative splicing events in SF3B1 knockdown group more or less frequently than control group were taken as the changed splicing pattern. The analysis revealed that exon skipping (SKIP) and cassette exons (MSKIP) were the most common molecular effect (p < 0.05) (Fig. 6b). In addition, SF3B1 knockdown barely affected other alternative splicing patterns, including retention of single (IR), multiple (MIR) introns, alternative exon ends (AE), alternative transcription start site (TSS) and alternative transcription termination site (TTS).

Analysis of different splicing events following SF3B1 knockdown. a Schematic illustration of different splicing events. b Exon skipping events predominate in MDA-MB-231 cells following SF3B1 knockdown
Discussion
SF3B1 mutations have been demonstrated to be driver events in breast cancer and SF3B1 levels significantly increased in ER-positive cells with endocrine resistance [14]. Overexpression of SF3B1 was also confirmed in prostate, hepatocellular and endometrial carcinoma [5, 15, 16]. Inconsistently, SF3B1 has been recently reported to harbor copy number (CN) loss in 71% chromophobe renal cell carcinoma and its CN loss was correlated with lower expression of SF3B1 [17]. In this study, SF3B1 is overexpressed in breast cancer samples and associated with lymph node metastasis. The discrepancy of SF3B1 expression in different cancers is probably attributed to the tissue type and different microenvironment.
We knocked down SF3B1 in ZR-75-30 cells which are ER-positive and MDA-MB-231 cells which represent triple negative breast cancer (TNBC), to observe the biological effect. The results showed that SF3B1 knockdown inhibited proliferation, invasion, migration and promoted apoptosis in both cell lines. Although SF3B1 mutation is more frequent in ER-positive breast cancer than TNBC, our study showed that the cancer-promoting effect of SF3B1 was more pronounced in TNBC than ER-positive breast cancer. Neelamraju’s study indicated that depletion of SF3B1 resulted in reduction of stem cell features in ER-positive breast cancer, but not in TNBC [18]. Neelamraju’s conclusions cannot provide a reasonable explanation for our results. In breast cancer, SF3B1 was reported to induce sensitivity to lack of serine, which was caused by the missplicing-associated downregulation of the phosphoglycerate dehydrogenase (PHGDH) involved in the early steps of L-serine synthesis [19]. It is worth noting that TNBC depends heavily on the serine synthesis signaling and some TNBC cases harbor amplification of PHGDH [20, 21]. Whether that means rare occurrence of SF3B1 mutations in TNBC is associated with suppressed serine synthesis pathway is unclear. It is interesting that our RNA sequencing in TNBC also identified serine metabolism pathway as a differentially expressed genes-enriched pathways upon SF3B1 knockdown. So, the intricate relationship between SF3B1 mutation, dysregulation of expression, serine synthesis and breast cancer subtype requires further research in the future.
SF3B1 mutation could lead to cryptic 3′splice site selection by alternative branch point sequence [22, 23]. The abnormal splice junctions probably result in nonsense-mediated decay (NMD) and mRNA down-regulation of affected genes. In this study, we identified a group of differentially expressed genes and aberrant spliced genes. However, whether these changes are related to mutation needs further research. Also in this study, our global RNA-sequencing analysis found that exon skipping is the primary consequence of SF3B1 knockdown. Our conclusion is supported by Wu’s results that exon skipping was also the major event in HeLa, HCT-116 and Rh18 cancer cells which were treated with SF3B1 inhibitor [24]. In contrast, Yoshimoto reported that intron retention was the main aberrant splicing event in HeLa cells treated with spliceostatin A, another SF3B1 inhibitor [25]. Interestingly, exon skipping was more prevalent than intron retention when Yoshimoto analyzed the data published by another group [25]. It is inferred that intron retention may be more prominent when normalized by the total number of the splicing events in cancer cells. Therefore, it is believable that in regard to differential splicing events following SF3B1 inhibition or knockdown, exon skipping is the most frequent.
The findings in this study may provide a rationale for the development of SF3B1 modulators for breast cancer therapy. In fact, SF3B1 modulators have progressed to preclinical and clinical trials in cancer therapy. E7107, a SF3B1 modulator that demonstrated remarkable antitumor efficacy in vivo, significantly improving the survival rates in diffuse malignant peritonealmesothelioma, lung cancer and colon carcinomas [26, 27]. Although visual impairment hindered further clinical development, the administration of E7107 in a Phase I trial stabilized the disease and resulted in partial response in cancer patients such as metastatic pancreatic cancer [28]. H3B-8800, an orally available SF3B inhibitor, preferentially induced death of spliceosome-mutant hematologic tumor cells at tolerated doses and currently entered a phase I clinical trial [29]. However, in breast cancer, there is no relevant report on the in vitro/vivo and clinical trials of SF3B1 modulator. Our study first confirmed the oncogene-like effect of SF3B1 in breast cancer and suggested the therapeutic potential of SF3B-targeting compounds against breast cancer. Further investigation is needed in the following plan to assess the potential antitumor effects of reducing SF3B1 activity by its modulator in breast cancer.
In conclusion, our findings demonstrate that SF3B1 expression is dramatically upregulated in the breast cancer and correlated with lymph node metastasis. SF3B1 knockdown in breast cancer can induce apoptosis and cell cycle arrest. SF3B1 knockdown can reduce the ability of cell proliferation, colony formation, invasion and migration. The exon skipping was the most common splicing event upon SF3B1 knockdown. Our study suggests that SF3B1 may be an important molecular target for cancer treatment of breast cancer.
References
El Marabti E, Younis I. The cancer spliceome: reprograming of alternative splicing in cancer. Front Mol Biosci. 2018;5:80.
Slansky JE, Spellman PT. Alternative splicing in tumors-a path to immunogenicity? N Engl J Med. 2019;380:877–80.
Maguire SL, Leonidou A, Wai P, Marchiò C, Ng CK, Sapino A, et al. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol. 2015;235:571–80.
Newell F, Kong Y, Wilmott JS, Johansson PA, Ferguson PM, Cui C, et al. Whole-genome landscape of mucosal melanoma reveals diverse drivers and therapeutic targets. Nat Commun. 2019;10:3163.
Jiménez-Vacas JM, Herrero-Aguayo V, Gómez-Gómez E, León-González AJ, Sáez-Martínez P, Alors-Pérez E, et al. Spliceosome component SF3B1 as novel prognostic biomarker and therapeutic target for prostate cancer. Transl Res. 2019;S1931–5244:30134–43.
Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–506.
Chang YS, Huang HD, Yeh KT, Chang JG. Genetic alterations in endometrial cancer by targeted next-generation sequencing. Exp Mol Pathol. 2016;100:8–12.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501.
Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Effenberger KA, Urabe VK, Prichard BE, Ghosh AK, Jurica MS. Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA. 2016;22:350–9.
Paolella BR, Gibson WJ, Urbanski LM, Alberta JA, Zack TI, Bandopadhayay P, et al. Copy-number and gene dependency analysis reveals partial copy loss of wild-type SF3B1 as a novel cancer vulnerability. Elife. 2017;6:e23268.
Mortera-Blanco T, Dimitriou M, Woll PS, Karimi M, Elvarsdottir E, Conte S, et al. SF3B1-initiating mutations in MDS-RSs target lymphomyeloid hematopoietic stem cells. Blood. 2017;130:881–90.
Zhang L, Zhou Y, Cheng C, Cui H, Cheng L, Kong P, et al. Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am J Hum Genet. 2015;96:597–611.
Gökmen-Polar Y, Neelamraju Y, Goswami CP, Gu X, Nallamothu G, Janga SC, et al. Expression levels of SF3B3 correlate with prognosis and endocrine resistance in estrogen receptor-positive breast cancer. Mod Pathol. 2015;28:677–85.
Guo S, Yang J, Wu M, Xiao G. Clinical value screening, prognostic significance and key pathway identification of miR-204-5p in endometrial carcinoma: A study based on the Cancer Genome Atlas (TCGA), and bioinformatics analysis. Pathol Res Pract. 2019;215:1003–111.
Hwang HM, Heo CK, Lee HJ, Kwak SS, Lim WH, Yoo JS, et al. Identification of anti-SF3B1 autoantibody as a diagnostic marker in patients with hepatocellular carcinoma. J Transl Med. 2018;16:177.
Ohashi R, Schraml P, Batavia A, Angori S, Simmler P, Rupp N, et al. Allele loss and reduced expression of CYCLOPS genes is a characteristic feature of chromophobe renal cell carcinoma. Transl Oncol. 2019;12:1131–7.
Neelamraju Y, Gonzalez-Perez A, Bhat-Nakshatri P, Nakshatri H, Janga SC. Mutational landscape of RNA-binding proteins in human cancers. RNA Biol. 2018;15:115–29.
Dalton WB, Helmenstine E, Walsh N, Gondek LP, Kelkar DS, Read A, et al. Hotspot SF3B1 mutations induce metabolic reprogramming and vulnerability to serine deprivation. J Clin Invest. 2019;130:4708–23.
Samanta D, Semenza GL. Serine synthesis helps hypoxic cancer stem cells regulate redox. Cancer Res. 2016;76:6458–62.
Zhang X, Bai W. Repression of phosphoglycerate dehydrogenase sensitizes triple-negative breast cancer to doxorubicin. Cancer Chemother Pharmacol. 2016;78:655–9.
DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, et al. Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in SF3B1-mutated cancers. PLoS Comput Biol. 2015;11:e1004105.
Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:10615.
Wu G, Fan L, Edmonson MN, Shaw T, Boggs K, Easton J, et al. Inhibition of SF3B1 by molecules targeting the spliceosome results in massive aberrant exon skipping. RNA. 2018;24:1056–66.
Yoshimoto R, Kaida D, Furuno M, Burroughs AM, Noma S, Suzuki H, et al. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. RNA. 2017;23:47–57.
Sciarrillo R, Wojtuszkiewicz A, El Hassouni B, Funel N, Gandellini P, Lagerweij T, et al. Splicing modulation as novel therapeutic strategy against diffuse malignant peritoneal mesothelioma. EBioMedicine. 2019;39:215–25.
Iwata M, Ozawa Y, Uenaka T, Shimizu H, Niijima J, et al. E7107, a new 7-urethane derivative of pladienolide D, displays curative effect against several human tumor xenografts [abstract 2989]. Proc Am Assoc Cancer Res. 2004;45:691.
Eskens FA, Ramos FJ, Burger H, O'Brien JP, Piera A, Piera A, et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res. 2013;19:6296–304.
Desterro J, Bak-Gordon P, Carmo-Fonseca M. Targeting mRNA processing as an anticancer strategy. Nat Rev Drug Discov. 2019. https://doi.org/10.1038/s41573-019-0042-3.
Acknowledgements
This work was supported by funding from the National Natural Science Foundation of China (81773150 to LZ.), Natural Science Foundation of Shanxi Province (201801D121339 to JL.), the Program for the Outstanding Innovative Teams of Higher Learning Institutions of Shanxi (OIT 2017 to LZ.), the Doctoral Start up Research Fund of Shanxi Medical University (03201508 to LZ.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
The present study was approved by the Ethics Committee of the Shanxi Medical University (approval No. 2018LL0115) and was conducted in accordance with the ethical standards. All procedures performed in studies involving human participants were in accordance the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration.
Informed consent
Informed consent was obtained from all individual participants included in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Zhang, L., Zhang, X., Zhang, H. et al. Knockdown of SF3B1 inhibits cell proliferation, invasion and migration triggering apoptosis in breast cancer via aberrant splicing. Breast Cancer 27, 464–476 (2020). https://doi.org/10.1007/s12282-020-01045-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12282-020-01045-8