
Abstract
Purpose of Review
This review examines how next-generation nucleic acid sequencing (NGS) is being used in the diagnosis of invasive fungal disease (IFD) and how well its implementation compares to PCR-based diagnosis. The comparison allows for the assessment of the advantages and potential limitations of NGS in the diagnosis of IFD.
Recent Findings
NGS is established as a method for sequencing of microbial genomes. It is increasingly being tested as a direct diagnostic method from various sample types including samples such as plasma, formalin fixed and paraffin embedded specimens, and bronchoalveolar lavage. In these cases, the methodology was generally able to identify the causative agents of IFD.
Summary
NGS-based methods will play an increasingly important role in diagnosis of IFD. The major limitation is currently cost, the need to standardise methods of nucleic acid isolation, and sequence analysis to enable broad uptake and application of the method.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It has been consistently reported that the incidence of fungal infections is increasing [5]. This applies to a range of animals in the environment, e.g. chytridiomycosis in amphibians [18] and white-nose disease in bats [60], as well as humans. The reason for this increase is complex and is dependent on the ecology of each fungal species. The main causal agents of invasive fungal disease (IFD) in humans are primarily opportunistic pathogens, e.g. Aspergillus, Fusarium, Rhizopus, or Scedosporium, that rarely cause disease in a healthy host [21]. The driver for the observed increase in IFD has been immunosuppression of the human host either by infection with a non-fungal primary pathogen, e.g. HIV, or through medical interventions such as solid organ and stem-cell transplantations [3, 61]. Post-tuberculosis infection, chronic pulmonary obstructive disorder (COPD), and long-term corticosteroid treatment are additional risk factors [54]. In the 1980s, the incidence of invasive disease caused by Candida increased due to acquired immune deficiency syndrome (AIDS) but it has been replaced by Pneumocystis and Cryptococcus infections in recent times driven by changes in patient care [31]. The majority of cases of candidiasis were previously caused by Candida albicans but there are now a wide variety of Candida species involved [48], including the recent emergence of Candida auris [57]. Similarly, the spectrum of fungi causing invasive mould diseases has been increasing, which has placed pressure on established diagnostic methodologies.
Although there are several effective treatments for IFD, there is still a persistent problem in diagnosis; early diagnosis is particularly important for the treatment of opportunistic mould infections [34, 52•]. For clinicians, the early symptoms of IFD are non-specific and empirical treatment for bacterial infections will often be administered. In patients with clearly defined risk factors for IFD, antifungal prophylaxis is administered but the mortality associated with break-through IFD remains high, 50–70% depending on the site of infection [47, 58]. Prophylaxis is not totally effective and break-throughs can occur; the combination of delayed diagnosis and drug-resistant fungi contributes to the high mortality rates. Since the 1980s, a number of diagnostic tests targeting fungus-derived molecules have been developed, including serum galactomannan [38], β-glucan testing [33], PCR [11••], and matrix-assisted laser desorption/ionisation-time of flight (MALDI-TOF) mass spectrometry [51]. These methods work by detecting fungal targets in blood or serum, and bronchoalveolar lavage (BAL). However, none of these has completely replaced culture and histology [30]. All methods can contribute valuable information for the clinician and have been gathered into the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group (EORTC) and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (MSG) (EORTC/MSG) guidelines for the definition for IFD; based on patient risk factors, traditional methods, and mycological factors, patients can be defined as having proven, probable, or possible IFD [13•].
The promise of molecular biology in IFD diagnosis has been boosted by advances in nucleic acid sequencing technology. The capacity to genotype or even sequence whole genomes and metagenomes in a short timeframe at an increasingly relatively low cost and to identify novel or rare pathogens has created new possibilities in the development of diagnostic strategies for IFD. This review will examine the applications and potential limitations of sequencing in the diagnosis of IFD.
Genome Sequencing in Medical Mycology
The topic of using whole genome sequencing (WGS) in medical mycology has previously been reviewed, so this review will focus on the applications of whole genome analyses to Candida, Cryptococcus, and Aspergillus [12••]. The first fungi to be sequenced were yeasts Saccharomyces cerevisiae, followed by Schizosaccharomyces pombe [66] with Neurospora crassa being the first mould to be sequenced [19]. These were all well-established model organisms used in genomic research for over 70 years, with a robust research community and molecular toolkit. The N. crassa genome had almost double the number of genes annotated compared to any yeast previously sequenced: 41% of its genome lacked homologs to known proteins, indicating yeasts are a poor proxy for all fungi [19]. The existence of reference genomes is an important consideration for the use of WGS in diagnostics. The process of fungal WGS follows a similar workflow irrespective of genus. First, genomicists construct one or more high-quality and high-level (chromosomal) reference genomes for widely available strains. Reference genomes can then be used to guide genome assembly and identify variants, allowing for even more cost-effective WGS studies focused on specific biological questions (resequencing of populations or strains). Initially, these reference sequencing projects were time consuming and expensive due to both high consumable and associated labour costs. However, they have become sufficiently accessible, for even smaller laboratories, since the introduction of the current generation of sequencing technology, frequently referred to as next-generation sequencing (NGS). Genomes of human pathogenic species from the genera Cryptococcus, Aspergillus, Candida, Pneumocystis, Histoplasma, Coccidioides, Mucor, Blastomyces, and Scedosporium have been sequenced, assembled, and published in the NCBI genome database [49], the primary repository for such information. The first C. albicans genome (strain SC5314), published in 2004, was a significant milestone in genomics as it required the development of new computational methods to overcome issues associated with heterozygous diploid species [32]. In 2005, the Aspergillus fumigatus AF293 genome sequence was first published [45]. This was compared with the genomes of Aspergillus nidulans and Aspergillus oryzae revealing low intra-genus amino acid identity, and a genomic capacity for heterothallic sexual reproduction in A. fumigatus [20]. The relatively low similarity within the genus Aspergillus has proven to be an issue in PCR-based diagnosis of IA and was examined in previous research [43]. The first genomes of Cryptococcus neoformans became publicly available in 2004–2005 [40]. These early WGS studies, together with complementary DNA sequencing (surveys the active genes, i.e. the transcriptome, and can help identify the genes in the genomes), identified 30 new genes putatively involved in synthesis of the polysaccharide capsule indispensable for C. neoformans virulence [40]. These early genomes provided a foundational resource for application of resequencing in medical mycology.
Monitoring Intraspecific Variation in Pathogenic Fungi
Using WGS for resequencing has proven to be valuable in the areas of fungal microevolution, resistance and virulence monitoring, and outbreak analysis (discussed in detail below). The primary studies in these areas required whole genome sequencing of target organisms and large-scale sequencing of specific gene targets to complement the initial WGS analyses. It is important to note that the quality of the determined variants depends on not only the quality of the reference genome assembly but also how representative it is for the resequenced strains. While short read NGS technologies can provide accurate identification of single base pair variants (SNVs), recent single molecule re-sequencing technologies, such as Oxford Nanopore, allow for improved detection of large structural variants, including copy number variants (CNVs) and pathogenicity islands [53•].
Microevolution in the Host
Whole genome sequencing of clinical C. albicans isolates, which had been sub-cultured both in vitro and in a murine model, has been used to characterise novel mutations that arise. One study [15] found microevolution to be driven primarily by amino-acid changing SNPs and short-tract loss-of-heterozygosity (LOH) events that lead to recombination-induced mutagenesis. WGS of C. albicans isolated from oral samples taken from healthy human hosts also found short-tract LOH events to be important in generating within-host variation. The high resolution of WGS revealed intra-sample heterogeneity, highlighting the importance of considering intra-host variability when comparing serial isolates [56].
Recent C. neoformans resequencing projects have characterised the adaptation of this fungus to the host environment. In these studies, isolates were serially sampled from patients over the course of infection, sequenced, and compared to identify microevolution events. Such studies have made significant findings. First, isolates recovered after relapse in cryptococcal meningitis patients are usually clonally related to the original infection [46•, 50•]. Second, aneuploidy of specific chromosomes (chromosome 12 in this case) [46•, 50•] and mutation of an AT-rich interaction domain protein may be important mechanisms of in-host adaptation [46•]. Third, non-sense mutations in DNA mismatch repair proteins can lead to a hypermutator state, accelerating the potential for microevolution [50•]. Sequencing data have also enabled identification of a genome amplification event that facilitates massive tandem gene amplification in response to environmental stimuli and drives microevolution [10]. Similar to C. neoformans, several recent A. fumigatus WGS projects have investigated in-host microevolution albeit with a greater focus on azole resistance [4•, 7, 26].
Antifungal Resistance
Resistance to antifungal drugs is the result of an arms-race between the populations of the hosts and the pathogen. Microevolution studies suggest that cyp51A SNPs conferring azole-resistance are selected during infection as are tandem repeats (TR120) in the cyp51A promoter selected during infection [26]. Resequencing of fluconazole-resistant isolates has implicated gain-of-function, Erg11 heterozygous and Erg3 homozygous mutations, and MDR1 promoter allele alterations in azole-resistance of C. albicans [8]. New experiments using resequencing have been used to identify mechanisms of resistance by genotyping strains in which genes conferring antifungal resistance have been deleted but resistance has been restored by artificial selection in the laboratory (experimental evolution). In the absence of Rgd1, an azole-resistance conferring gene, exposure to azoles induced amplification of several chromosomal regions as identified by WGS. Overexpression of a transporter gene, NPR2 was found to confer resistance [44]. A similar study exploring the effects of medium-chain fatty acids found susceptibility to be associated with trisomy of chromosome 7 [42].
WGS of C. albicans and S. cerevisiae strains experimentally evolved for resistance to co-treatment with azoles and inhibitors of either Hsp90 or calcineurin inhibitors revealed diverse resistance mechanisms including extensive aneuploidies and mutations in genes encoding drug targets; transcriptional regulators of multidrug transporters and ergosterol biosynthesis enzymes; and Lcb1, a regulator of sphingolipid biosynthesis and extensive aneuploidies [28]. Resequencing has been used to genotype experimentally evolved azole-resistant A. fumigatus isolates relative to their isogenic parental strains. Both medical and non-medical triazole fungicides have been used in experimental evolution studies. Variants contributing to medical triazole induced resistance includes mutations in erg11A (cyp51A), multidrug transporters, erg25, and HMG-CoA reductase [41]. Agricultural fungicides induced cross-resistance to medical triazoles with mutations also seen in cyp51A and HMG-CoA reductase [68•].
Outbreak and Virulence Analysis
Cryptococcus gattii is less common than C. neoformans but can infect immunocompetent individuals. While once considered endemic to tropical and subtropical environments, C. gattii outbreaks in the Pacific north west of the USA precipitated the need for phylogenetic studies to help identify outbreak origin. The clonal nature of C. gattii sublineages impeded the ability of multilocus sequence typing (MLST) to resolve variation [23] but resequencing of 118 genomes managed to identify South America as the probable origin of Pacific north west lineages [16].
Resequencing of 56 C. neoformans strains identified 40 genes as putatively associated with human survival, immunologic response or clinical parameters. Using the 17 available KN99α gene deletion strains for these candidate genes—six (35%) were found to directly influence survival of mouse models: three increased and three decreased survival, four of which had not previously been identified [22••]. In recent years, SNP-based phylogenies have shown mucosal and bloodstream C. albicans isolates are organised into separate clades [6]. Resequencing and phenotyping of two clinical isolates of variable pathogenic potential indicate major differentiating genetic variants are located in genes associated with biofilm production and first-line host barriers while they vary genetically in a manner that correlates to isolate-specific phenotypic differences [9].
Sequencing in the Diagnosis of Invasive Fungal Disease
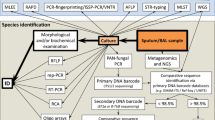
Sequence analysis of fungal DNA from patient samples is well established and being established depending on the techniques being employed (Fig. 1). Sequencing of PCR products has been common practice in the identification of organisms causing IFD. This has usually involved isolation of DNA from formalin fixed paraffin embedded (FFPE) tissue samples or samples of mycelium. This practice usually involves PCR amplification of a variable ribosomal region, usually with ITS-targeting primers, and Sanger sequencing, the sequence is compared to existing sequences using a sequence similarity algorithm (https://blast.ncbi.nlm.nih.gov/Blast.cgi). In contrast, qPCR assays rely on existing sequence data for the design of oligonucleotide primers and probes for targeted amplification of IFD causing pathogenic fungi. The quality and diversity of existing sequence data usually dictates the level of specificity achievable with qPCR assays.

Schematic diagram indicating the strategies for isolation of DNA from different samples and the subsequent options for processing the DNA to identify the causes of invasive fungal disease in patient samples. 1The additional steps include separation of blood samples into cell-free (serum or plasma) or white cell fractions, xylene and ethanol treatment for FFPE, proteinase K treatment for tissue, and lyticase or bead beating to release DNA from fungal spores or hyphae. 2The ITS 4/5 primer set [62] has been used extensively in the identification of fungi due to the large number of sequences submitted to the DNA databases. 3High-resolution meltcurve (HRM) analysis. 4Multiplex qPCR can have multiple probes to allows identification of fungal genus or species and detect the presence of drug resistance alleles
Recent NGS protocols create the possibility to perform targeted sequencing of pan-fungal PCR products from a sample or non-targeted sequencing of all DNA present in the sample allowing for the reconstruction of the community of organisms present in the sample. In a study of three cases of individuals at-risk for IA, NGS was used to sequence all the DNA from BAL samples. NGS was able to identify A. fumigatus in all three cases and was the only positive diagnostic method in one case [27••]. A further study examined the use of NGS in serum samples from nine patients with proven IFD [29••]. In this study, NGS identified a fungal pathogen in seven of the nine cases. Pulmonary scedosporiosis was diagnosed with the aid of NGS, where DNA from BAL and NGS results were confirmed by histology [67••]. An important aspect of using any diagnostic method is sample type and cell-free DNA in serum/plasma is an attractive target that has performed well in PCR-based studies [39, 64]. A trial of NGS to diagnose IFD in paediatric patients using plasma identified 4 of 6 (66.67%) cases of proven fungal disease. The 2 of 6 (33.33%) cases that were not identified appeared to have non-invasive infections [1••]. These cases highlight some of the advantages of NGS in diagnosis; it can detect any potential pathogen in a sample and the surrounding microbial community. It can be performed on any sample type from which DNA or RNA can be isolated including the usual diagnostic samples types, e.g. blood, serum, BAL, and FFPE.
The use of NGS avoids the need to develop pathogen specific assays, a major technical issue for qPCR assays. The fungal databases do not contain sufficient information to screen primers and probes to ensure specificity. Even if the databases were complete the taxonomic issues within genera such as Aspergillus make it difficult to develop a pan-Aspergillus PCR [43]. NGS can provide species level identification from the raw data generated, which avoids the need for a secondary identification step and allows targeted treatment of the pathogen. The case study by Hong et al. identified Aspergillus lentulus from a clinical sample whereas other methods would identify A. fumigatus [29••]. This is critical since Aspergillus species vary considerably in their responses to antifungal drugs [59]. The turnaround time for NGS has been decreasing as the methodology has been refined and now results can be expected within 48-72 h, an acceptable timeframe for a species-level and antifungal resistance identification.
Potential Limitations of NGS in IFD Diagnosis
Although the workflow is very similar to PCR-based assays with relation to sample type and DNA isolation (Fig. 1), there are some differences and challenges that should be recognised.
The key problem for nucleic acid diagnostic assays is having sufficient nucleic acid in the sample. In the standardisation of qPCR assays, it was found that the key factor affecting performance was the DNA isolation methodology [63••]. This is the input for any PCR or sequencing-based assay and is a more important consideration than specific amplification target or platform. Fungal DNA occurs at very low levels, near the limit of detection for qPCR, in blood and serum creating a potential problem for NGS-based approaches. At these low levels of target, the DNA may be indistinguishable from contamination, which is an issue in microbiome studies with smaller samples [14] and has even led to conflicting accounts of a human placental microbiome [36]. Single molecule sequencing technologies are meant to, ultimately, overcome this barrier but the laboratory protocols are not yet optimised and inputs in the range of 10–100 s of ng of DNA are still required while significant sequencing consumables are also ‘wasted’ on non-target host DNA. Indeed, another question is whether NGS is sufficiently sensitive to detect pathogens that are in low abundance since the method can be influenced by the abundance of competing DNA in the sample (see these reviews for further discussion on these technical limitations [24•, 25•]). A third challenge is that the majority of diagnostic strategies for IFD suggest twice weekly sampling and analysis to ensure that the pathogen is detected [13•]; this may not be cost-efficient for NGS yet due to cost and time considerations. Another related issue is the use of antifungal prophylaxis, which reduces the amount of diagnostic targets in the host and has a pronounced effect on the performance of qPCR assays which would also affect NGS [11••].
A qPCR assay gives a defined result allowing immediate interpretation of a positive or negative result. It is generally accepted that the strength of qPCR is in its ability to exclude the presence of IFD rather than to detect the pathogen [11••]. In a diagnostic platform utilising several strands of information, this type of test can be beneficial but there is still insufficient information about the interactions between IFD-causing fungi and the host to enable certainty when interpreting what a positive sample means. By comparison to PCR-based assays, direct NGS from a sample can yield a wealth of microbial information in terms of the number and identity of organisms present in a sample. This wealth of information creates a challenge in the scientific and medical interpretation of the data, e.g. what should the threshold for an IFD be? Which organisms are significant? Indeed, the data from NGS can overwhelm clinicians with less than relevant data [37].
Sample origin and type is another important consideration. From a serum sample, it might be deduced that a positive result for the presence of a pathogen may be significant since the DNA from pathogens is in such low abundance in that sample type. Analysing BAL samples may be more challenging since the lung has a transient microbiome that consists of a variety of fungi, bacteria, parasites, and viruses [17]. The use of qPCR from BAL raised questions about the interpretation of a positive result, leading to implementation of thresholds and distinguishing between colonisation and infection [35, 55]. The data from NGS would be more complex and would require the definition of stringent thresholds for classifying cases for IFD. There is still insufficient information about the lung microbiome especially in individuals at-risk for IFD to define such thresholds. Increasing data may allow the definition of what a dysbiosis associated with IFD might look like; this would remove the pressure from detecting a specific pathogen and focus on microbial population dynamics.
Conclusions
It has taken almost 20 years for qPCR assays to gain broad acceptance in the diagnosis of IFD and this required significant evidence of comparable performance to other diagnostic assays [65]. This was greatly aided by the use of meta-analyses to identify the variation in assay performance [2, 11••] and led to community efforts to standardise PCR-based methodologies such as the Fungal PCR Initiative (https://www.isham.org/working-groups/european-aspergillus-pcr-initiative-eapcri). Multicentre analysis of standardised samples, optimised DNA isolation protocols for different sample types, standardised pipelines for data analysis, and guidelines for the interpretation of the data would bring NGS into line with other diagnostic methods for IFD. NGS in the diagnosis of IFD could be the future but there should be a community approach to developing standardised protocols to get the best from this technology.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Armstrong AE, Rossoff J, Hollemon D, Hong DK, Muller WJ, Chaudhury S. Cell-free DNA next-generation sequencing successfully detects infectious pathogens in pediatric oncology and hematopoietic stem cell transplant patients at risk for invasive fungal disease. Pediatr Blood Cancer. 2019;66(7):e27734. https://doi.org/10.1002/pbc.27734Use of NGS to detect cell-free DNA from pathogenic microbes in plasma.
Arvanitis M, Ziakas PD, Zacharioudakis IM, Zervou FN, Caliendo AM, Mylonakis E. PCR in diagnosis of invasive aspergillosis: a meta-analysis of diagnostic performance. J Clin Microbiol. 2014;52(10):3731–42. https://doi.org/10.1128/JCM.01365-14.
Badiee P, Kordbacheh P, Alborzi A, Malekhoseini S, Ramzi M, Mirhendi H, et al. Study on invasive fungal infections in immunocompromised patients to present a suitable early diagnostic procedure. Int J Infect Dis. 2009;13(1):97–102. https://doi.org/10.1016/j.ijid.2008.04.011.
• Ballard E, Melchers WJG, Zoll J, Brown AJP, Verweij PE, Warris A. In-host microevolution of Aspergillus fumigatus: a phenotypic and genotypic analysis. Fungal Genet Biol. 2018;113:1–13. https://doi.org/10.1016/j.fgb.2018.02.003Uses sequencing (comparative genomics) to examine if host microenvironments lead to a selection pressure driving adaptation to the host inAspergillus fumigatus.
Bongomin F, Gago S, Oladele RO, Denning DW. Global and multi-national prevalence of fungal diseases-estimate precision. J Fungi (Basel). 2017;3(4). https://doi.org/10.3390/jof3040057.
Bradford LL, Chibucos MC, Ma B, Bruno V, Ravel J. Vaginal Candida spp. genomes from women with vulvovaginal candidiasis. Pathog Dis. 2017;75(6). https://doi.org/10.1093/femspd/ftx061.
Camps SM, Dutilh BE, Arendrup MC, Rijs AJ, Snelders E, Huynen MA, et al. Discovery of a HapE mutation that causes azole resistance in Aspergillus fumigatus through whole genome sequencing and sexual crossing. PLoS One. 2012;7(11):e50034. https://doi.org/10.1371/journal.pone.0050034.
Castanheira M, Deshpande LM, Davis AP, Rhomberg PR, Pfaller MA. Monitoring antifungal resistance in a global collection of invasive yeasts and molds: application of CLSI epidemiological cutoff values and whole-genome sequencing analysis for detection of azole resistance in Candida albicans. Antimicrob Agents Chemother. 2017;61(10). https://doi.org/10.1128/AAC.00906-17.
Cavalieri D, Di Paola M, Rizzetto L, Tocci N, De Filippo C, Lionetti P, et al. Genomic and phenotypic variation in morphogenetic networks of two Candida albicans isolates subtends their different pathogenic potential. Front Immunol. 2017;8:1997. https://doi.org/10.3389/fimmu.2017.01997.
Chow EW, Morrow CA, Djordjevic JT, Wood IA, Fraser JA. Microevolution of Cryptococcus neoformans driven by massive tandem gene amplification. Mol Biol Evol. 2012;29(8):1987–2000. https://doi.org/10.1093/molbev/mss066.
•• Cruciani M, Mengoli C, Loeffler J, Donnelly P, Barnes R, Jones BL, et al. Polymerase chain reaction blood tests for the diagnosis of invasive aspergillosis in immunocompromised people. Cochrane Database Syst Rev. 2015;(10):CD009551. https://doi.org/10.1002/14651858.CD009551.pub3Comprehensive meta-analysis of the application of PCR for the diagnosis of invasive aspergillosis in at-risk patients, recently updated to show current state of the field.
•• Cuomo CA. Harnessing whole genome sequencing in medical mycology. Curr Fungal Infect Rep. 2017;11(2):52–9. https://doi.org/10.1007/s12281-017-0276-7Comprehensive review of application of whole genome sequencing to student clinically important fungal pathogens, complementary to the current review.
• De Pauw B, Walsh TJ, Donnelly JP, Stevens DA, Edwards JE, Calandra T, et al. Revised definitions of invasive fungal disease from the European Organization for Research and Treatment of Cancer/Invasive Fungal Infections Cooperative Group and the National Institute of Allergy and Infectious Diseases Mycoses Study Group (EORTC/MSG) Consensus Group. Clin Infect Dis. 2008;46(12):1813–21. https://doi.org/10.1086/588660Describes the evidence necessary to deliver a diagnosis of IFD in at-risk patients. Highlights the challenges associated with diagnosing these diseases.
Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 2019;27(2):105–17. https://doi.org/10.1016/j.tim.2018.11.003.
Ene IV, Farrer RA, Hirakawa MP, Agwamba K, Cuomo CA, Bennett RJ. Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc Natl Acad Sci U S A. 2018;115(37):E8688–E97. https://doi.org/10.1073/pnas.1806002115.
Engelthaler DM, Hicks ND, Gillece JD, Roe CC, Schupp JM, Driebe EM, et al. Cryptococcus gattii in North American Pacific Northwest: whole-population genome analysis provides insights into species evolution and dispersal. MBio. 2014;5(4):e01464–14. https://doi.org/10.1128/mBio.01464-14.
Faner R, Sibila O, Agusti A, Bernasconi E, Chalmers JD, Huffnagle GB, et al. The microbiome in respiratory medicine: current challenges and future perspectives. Eur Respir J. 2017;49(4). https://doi.org/10.1183/13993003.02086-2016.
Fisher MC, Garner TW, Walker SF. Global emergence of Batrachochytrium dendrobatidis and amphibian chytridiomycosis in space, time, and host. Annu Rev Microbiol. 2009;63:291–310. https://doi.org/10.1146/annurev.micro.091208.073435.
Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature. 2003;422(6934):859–68. https://doi.org/10.1038/nature01554.
Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2005;438(7071):1105–15. https://doi.org/10.1038/nature04341.
Garcia-Vidal C, Viasus D, Carratala J. Pathogenesis of invasive fungal infections. Curr Opin Infect Dis. 2013;26(3):270–6. https://doi.org/10.1097/QCO.0b013e32835fb920.
•• Gerstein AC, Jackson KM, McDonald TR, Wang Y, Lueck BD, Bohjanen S, et al. Identification of pathogen genomic differences that impact human immune response and disease during Cryptococcus neoformans Infection. MBio. 2019;10(4). https://doi.org/10.1128/mBio.01440-19Identification of virulence factors for human infection through whole genome comparisons.
Gillece JD, Schupp JM, Balajee SA, Harris J, Pearson T, Yan Y, et al. Whole genome sequence analysis of Cryptococcus gattii from the Pacific Northwest reveals unexpected diversity. PLoS One. 2011;6(12):e28550. https://doi.org/10.1371/journal.pone.0028550.
• Goldberg B, Sichtig H, Geyer C, Ledeboer N, Weinstock GM. Making the leap from research laboratory to clinic: challenges and opportunities for next-generation sequencing in infectious disease diagnostics. MBio. 2015;6(6):e01888–15. https://doi.org/10.1128/mBio.01888-15Short review detailing some of the challenges faced in adapting NGS for clinical use, especially for diagnostics.
• Greninger AL. The challenge of diagnostic metagenomics. Expert Rev Mol Diagn. 2018;18(7):605–15. https://doi.org/10.1080/14737159.2018.1487292Review examining the use of metagenomics in clinical applications, another perspective on the potential challenges associated with this methodology.
Hare RK, Gertsen JB, Astvad KMT, Degn KB, Lokke A, Stegger M, et al. In vivo selection of a unique tandem repeat mediated azole resistance mechanism (TR120) in Aspergillus fumigatus cyp51A, Denmark. Emerg Infect Dis. 2019;25(3):577–80. https://doi.org/10.3201/eid2503.180297.
•• He BC, Liu LL, Chen BL, Zhang F, Su X. The application of next-generation sequencing in diagnosing invasive pulmonary aspergillosis: three case reports. Am J Transl Res. 2019;11(4):2532–9 Use of NGS in assisting with the diagnsosis of IA, important demonstration of the use of the method.
Hill JA, Ammar R, Torti D, Nislow C, Cowen LE. Genetic and genomic architecture of the evolution of resistance to antifungal drug combinations. PLoS Genet. 2013;9(4):e1003390. https://doi.org/10.1371/journal.pgen.1003390.
•• Hong DK, Blauwkamp TA, Kertesz M, Bercovici S, Truong C, Banaei N. Liquid biopsy for infectious diseases: sequencing of cell-free plasma to detect pathogen DNA in patients with invasive fungal disease. Diagn Microbiol Infect Dis. 2018;92(3):210–3. https://doi.org/10.1016/j.diagmicrobio.2018.06.009A study that demonstrates a feasible diagnostic pathway using NGS in diagnosis of IFD.
Hope WW, Walsh TJ, Denning DW. Laboratory diagnosis of invasive aspergillosis. Lancet Infect Dis. 2005;5(10):609–22. https://doi.org/10.1016/S1473-3099(05)70238-3.
Ives NJ, Gazzard BG, Easterbrook PJ. The changing pattern of AIDS-defining illnesses with the introduction of highly active antiretroviral therapy (HAART)in a London clinic. J Infect. 2001;42(2):134–9. https://doi.org/10.1053/jinf.2001.0810.
Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, et al. The diploid genome sequence of Candida albicans. Proc Natl Acad Sci U S A. 2004;101(19):7329–34. https://doi.org/10.1073/pnas.0401648101.
Karageorgopoulos DE, Vouloumanou EK. Ntziora F, Michalopoulos A, Rafailidis PI, Falagas ME. beta-D-glucan assay for the diagnosis of invasive fungal infections: a meta-analysis. Clin Infect Dis. 2011;52(6):750–70. https://doi.org/10.1093/cid/ciq206.
Lamoth F, Calandra T. Early diagnosis of invasive mould infections and disease. J Antimicrob Chemother. 2017;72(suppl_1):i19–28. https://doi.org/10.1093/jac/dkx030.
Langford S, Trubiano JA, Saxon S, Spelman D, Morrissey CO. Mucormycete infection or colonisation: experience of an Australian tertiary referral centre. Mycoses. 2016;59(5):291–5. https://doi.org/10.1111/myc.12467.
Lauder AP, Roche AM, Sherrill-Mix S, Bailey A, Laughlin AL, Bittinger K, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome. 2016;4(1):29. https://doi.org/10.1186/s40168-016-0172-3.
Lecuit M, Eloit M. The potential of whole genome NGS for infectious disease diagnosis. Expert Rev Mol Diagn. 2015;15(12):1517–9. https://doi.org/10.1586/14737159.2015.1111140.
Leeflang MM, Debets-Ossenkopp YJ, Wang J, Visser CE, Scholten RJ, Hooft L, et al. Galactomannan detection for invasive aspergillosis in immunocompromised patients. Cochrane Database Syst Rev. 2015;12:CD007394. https://doi.org/10.1002/14651858.CD007394.pub2.
Loeffler J, Mengoli C, Springer J, Bretagne S, Cuenca-Estrella M, Klingspor L, et al. Analytical comparison of in vitro-spiked human serum and plasma for PCR-based detection of Aspergillus fumigatus DNA: a study by the European Aspergillus PCR Initiative. J Clin Microbiol. 2015;53(9):2838–45. https://doi.org/10.1128/JCM.00906-15.
Loftus BJ, Fung E, Roncaglia P, Rowley D, Amedeo P, Bruno D, et al. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science. 2005;307(5713):1321–4. https://doi.org/10.1126/science.1103773.
Losada L, Sugui JA, Eckhaus MA, Chang YC, Mounaud S, Figat A, et al. Genetic analysis using an isogenic mating pair of Aspergillus fumigatus identifies azole resistance genes and lack of MAT Locus’s role in virulence. PLoS Pathog. 2015;11(4):e1004834. https://doi.org/10.1371/journal.ppat.1004834.
Ma Q, Ola M, Iracane E, Butler G. Susceptibility to medium-chain fatty acids is associated with trisomy of chromosome 7 in Candida albicans. mSphere. 2019;4(3). https://doi.org/10.1128/mSphere.00402-19.
Morton CO, White PL, Barnes RA, Klingspor L, Cuenca-Estrella M, Lagrou K, et al. Determining the analytical specificity of PCR-based assays for the diagnosis of IA: What is Aspergillus? Med Mycol. 2017;55(4):402–13. https://doi.org/10.1093/mmy/myw093.
Mount HO, Revie NM, Todd RT, Anstett K, Collins C, Costanzo M, et al. Global analysis of genetic circuitry and adaptive mechanisms enabling resistance to the azole antifungal drugs. PLoS Genet. 2018;14(4):e1007319. https://doi.org/10.1371/journal.pgen.1007319.
Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature. 2005;438(7071):1151–6. https://doi.org/10.1038/nature04332.
• Ormerod KL, Morrow CA, Chow EW, Lee IR, Arras SD, Schirra HJ, et al. Comparative genomics of serial isolates of cryptococcus neoformans reveals gene associated with carbon utilization and virulence. G3 (Bethesda). 2013;3(4):675–86. https://doi.org/10.1534/g3.113.005660Use of sequencing to examine potential role of in-host evolution to the persistance and development of cryptococcal meningitis.
Pagano L, Mayor S. Invasive fungal infections in high-risk patients: report from TIMM-8 2017. Future Sci OA. 2018;4(6):FSO307. https://doi.org/10.4155/fsoa-2018-0019.
Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20(1):133–63. https://doi.org/10.1128/CMR.00029-06.
Prakash PY, Irinyi L, Halliday C, Chen S, Robert V, Meyer W. Online databases for taxonomy and identification of pathogenic fungi and proposal for a cloud-based dynamic data network platform. J Clin Microbiol. 2017;55(4):1011–24. https://doi.org/10.1128/JCM.02084-16.
• Rhodes J, Beale MA, Vanhove M, Jarvis JN, Kannambath S, Simpson JA, et al. A population genomics approach to assessing the genetic basis of within-host microevolution underlying recurrent cryptococcal meningitis infection. G3 (Bethesda). 2017;7(4):1165–76. https://doi.org/10.1534/g3.116.037499Use of sequencing to provide insights into the epidemiology of cryptococcal meningitis in HIV/AIDS patients.
Sanguinetti M, Posteraro B. Identification of molds by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol. 2017;55(2):369–79. https://doi.org/10.1128/JCM.01640-16.
• Sanguinetti M, Posteraro B, Beigelman-Aubry C, Lamoth F, Dunet V, Slavin M, et al. Diagnosis and treatment of invasive fungal infections: looking ahead. J Antimicrob Chemother. 2019;74(Supplement_2):ii27–37. https://doi.org/10.1093/jac/dkz041Wide-rangining review covering recent developments and challenges in the diagnosis and treatment of invasive fungal diseases.
• Schmidt K, Mwaigwisya S, Crossman LC, Doumith M, Munroe D, Pires C, et al. Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J Antimicrob Chemother. 2017;72(1):104–14. https://doi.org/10.1093/jac/dkw397Use of the Nanopore MinIon platform to identify pathogens in urine, this is potentially the future of sequencing technology in diagnostics. The device is suitable for any location and can provide relatively rapid results.
Segal BH. Aspergillosis. N Engl J Med. 2009;360(18):1870–84. https://doi.org/10.1056/NEJMra0808853.
Shahi M, Ayatollahi Mousavi SA, Nabili M, Aliyali M, Khodavaisy S, Badali H. Aspergillus colonization in patients with chronic obstructive pulmonary disease. Curr Med Mycol. 2015;1(3):45–51. https://doi.org/10.18869/acadpub.cmm.1.3.45.
Sitterle E, Maufrais C, Sertour N, Palayret M, d'Enfert C, Bougnoux ME. Within-host genomic diversity of Candida albicans in healthy carriers. Sci Rep. 2019;9(1):2563. https://doi.org/10.1038/s41598-019-38768-4.
Spivak ES, Hanson KE. Candida auris: an emerging fungal pathogen. J Clin Microbiol. 2018;56(2). https://doi.org/10.1128/JCM.01588-17.
Valentine JC, Morrissey CO, Tacey MA, Liew D, Patil S, Peleg AY, et al. A population-based analysis of invasive fungal disease in haematology-oncology patients using data linkage of state-wide registries and administrative databases: 2005 - 2016. BMC Infect Dis. 2019;19(1):274. https://doi.org/10.1186/s12879-019-3901-y.
Van Der Linden JW, Warris A, Verweij PE. Aspergillus species intrinsically resistant to antifungal agents. Med Mycol. 2011;49(Suppl 1):S82–9. https://doi.org/10.3109/13693786.2010.499916.
Warnecke L, Turner JM, Bollinger TK, Lorch JM, Misra V, Cryan PM, et al. Inoculation of bats with European Geomyces destructans supports the novel pathogen hypothesis for the origin of white-nose syndrome. Proc Natl Acad Sci U S A. 2012;109(18):6999–7003. https://doi.org/10.1073/pnas.1200374109.
Webb BJ, Ferraro JP, Rea S, Kaufusi S, Goodman BE, Spalding J. Epidemiology and clinical features of invasive fungal infection in a US Health Care Network. Open Forum Infect Dis. 2018;5(8):ofy187. https://doi.org/10.1093/ofid/ofy187.
White TJ, Bruns T, Lee SI, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR protocols: a guide to methods and applications. New York: Academic Press, Inc.; 1990.
•• White PL, Bretagne S, Klingspor L, Melchers WJ, McCulloch E, Schulz B, et al. Aspergillus PCR: one step closer to standardization. J Clin Microbiol. 2010;48(4):1231–40. https://doi.org/10.1128/JCM.01767-09Shows the importance of standardisation and the rigour necessary in the development of molecualr diagnostics for IA.
White PL, Barnes RA, Springer J, Klingspor L, Cuenca-Estrella M, Morton CO, et al. Clinical performance of Aspergillus PCR for testing serum and plasma: a study by the European Aspergillus PCR initiative. J Clin Microbiol. 2015;53(9):2832–7. https://doi.org/10.1128/JCM.00905-15.
White PL, Wingard JR, Bretagne S, Loffler J, Patterson TF, Slavin MA, et al. Aspergillus polymerase chain reaction: systematic review of evidence for clinical use in comparison with antigen testing. Clin Infect Dis. 2015;61(8):1293–303. https://doi.org/10.1093/cid/civ507.
Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, et al. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415(6874):871–80. https://doi.org/10.1038/nature724.
•• Xiao W, Han P, Xu Z, Huang M. Pulmonary scedosporiosis in a patient with acute hematopoietic failure: diagnosis aided by next-generation sequencing. Int J Infect Dis. 2019;85:114–6. https://doi.org/10.1016/j.ijid.2019.05.033Combines use of traditional diagnostics with NGS to diagnose a fungal infection.
• Zhang J, van den Heuvel J, Debets AJM, Verweij PE, Melchers WJG, Zwaan BJ, et al. Evolution of cross-resistance to medical triazoles in Aspergillus fumigatus through selection pressure of environmental fungicides. Proc Biol Sci. 2017;284(1863). https://doi.org/10.1098/rspb.2017.0635Investigation into the evolution of azole resistance inA. fumigatus, examining the hypothesis that agricultural use of azoles has driven resistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Sam El-Kamand, Alexie Papanicolaou and C. Oliver Morton declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Advances in Diagnosis of Invasive Fungal Infections
Rights and permissions
About this article

Cite this article
El-Kamand, S., Papanicolaou, A. & Morton, C.O. The Use of Whole Genome and Next-Generation Sequencing in the Diagnosis of Invasive Fungal Disease. Curr Fungal Infect Rep 13, 284–291 (2019). https://doi.org/10.1007/s12281-019-00363-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12281-019-00363-5