
Abstract
Bone undergoes continuous remodeling by a coupled action between osteoblasts and osteoclasts. During osteoporosis, osteoclast activity is often elevated leading to increased bone destruction. Hence, osteoclasts are deemed as potential therapeutic targets to alleviate bone loss. Ellagic acid (EA) is a polyphenol reported to possess anticancer, antioxidant and anti-inflammatory properties. However, its effects on osteoclast formation and function have not yet been examined. Here, we explored the effects of EA on RANKL-induced osteoclast differentiation in RAW264.7 murine macrophages (in vitro) and human CD14+monocytes (ex vivo). EA dose-dependently attenuated RANKL-induced TRAP+ osteoclast formation in osteoclast progenitors with maximal inhibition seen at 1 µM concentration without cytotoxicity. Moreover, owing to perturbed osteoclastogenesis, EA disrupted actin ring formation and bone resorptive function of osteoclasts. Analysis of the underlying molecular mechanisms revealed that EA suppressed the phosphorylation and activation of the p38 MAP kinase pathway which subsequently impaired the RANKL-induced differentiation of osteoclast progenitors. Taken together, these novel results indicate that EA alleviates osteoclastogenesis by suppressing the p38 signaling pathway downstream of RANKL and exerts inhibitory effects on bone resorption and actin ring formation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoporosis occurs due to an imbalance in bone remodeling owing to increased bone resorption and decreased bone formation resulting in excessive bone loss (Rachner et al. 2011). Factors accounting for osteoporosis include hormonal imbalance, ageing, or medications such as glucocorticoids. Osteoclasts, the sole bone resorptive cells of the body, arise from the hematopoietic cells of monocyte/macrophage lineage (Charles and Aliprantis 2014). Available evidence suggests that osteoclast formation or activity is highly elevated during bone loss related diseases such as osteoporosis, periodontitis, rheumatoid arthritis and cancer metastases to bone (Helfrich 2003, 2005; Wada et al. 2006). Current drugs prescribed for the treatment of bone loss include bisphosphonates, parathyroid hormone, estrogen and denosumab (Rodan and Martin 2000; Chen and Sambrook 2012; Whitaker et al. 2012; Augustine and Horwitz 2013). These therapeutics act by inhibiting bone loss triggered by osteoclast activity. Considering the fact that osteoclast activity is indispensable for bone resorption, targeting these cells is a fruitful approach for the treatment of osteoporosis.
Receptor activator of nuclear factor kappa B (RANK), its ligand RANKL and the decoy receptor of RANKL, osteoprotegerin (OPG) are crucial and key regulators of osteoclast formation (Walsh and Choi 2014). RANKL is expressed in membrane-bound form by osteoblasts, and osteoclast progenitors expressing its receptor RANK bind to RANKL to trigger the downstream signaling cascade pivotal for osteoclastogenesis (Hofbauer et al. 2001). During certain pathological conditions such as cancer metastases to bone and rheumatoid arthritis, RANKL is secreted in soluble form and is similarly active as its membrane-bound counterpart (Hofbauer et al. 2001). Earlier reports indicate the crucial role of RANKL in osteoclast formation, as mice devoid of RANKL not only develop severe osteopetrosis but they completely lack osteoclasts (Boyce and Xing 2008). Binding of RANKL to its receptor RANK activates the downstream signaling cascade that involves trimerization of adaptor protein TRAF6 resulting in activation of the NF-kB and MAP kinase pathways, and leads to expression of osteoclast-specific genes and osteoclast differentiation (Wada et al. 2006).
Polyphenols have been shown to possess various health-promoting effects (Marquardt and Watson 2014). Ellagic acid (EA, Fig. 1a) is a major non-flavonoid polyphenol found in numerous fruits including pomegranates, pecans, and raspberries, and exerts various pharmacological effects such as anti-cancer, antioxidant, anti-inflammatory and bactericidal properties (Usta et al. 2013). Accumulating lines of evidence suggest that phytochemicals with potent antioxidant and anti-inflammatory properties have bone protective effects and suppress bone resorption, resulting in greater bone strength (Shen et al. 2012). Hence, we tested the molecular effects of EA on RANKL-induced osteoclast formation. Here, we report that EA inhibits RANKL-induced osteoclastogenesis in RAW264.7 macrophages, in vitro and human CD14+ monocytes, ex vivo at a very low concentration. EA attenuated RANKL-induced p38 activation in macrophages without affecting the NF-kB pathway. On the other hand, EA did not affect the survival of mature osteoclasts indicating its specific inhibitory effects on osteoclast formation. Our results indicate that EA may be a novel anti-osteoporotic agent.

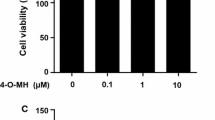
Effects of EA on the viability of murine RAW264.7 macrophages and human CD14+ monocytes. a Molecular structure of EA. b Cell viability of EA-treated RAW264.7 macrophages or c CD14+ monocytes. Cells were treated with indicated concentrations of EA for 48 h and cell viability was measured by alamar blue assay. Data are expressed as mean ± SD percent of control and are representative of three independent experiments
Materials and methods
Materials
Dulbecco’s Modified Eagle Medium (DMEM), α-MEM and heat-inactivated fetal bovine serum (FBS) were obtained from GIBCO (Grand Island, NY) and Amersham (Little Chalfont, UK), respectively. Antibiotic–antimycotic solution was supplied by Highveld Biological (Johannesburg, South Africa). EA, phalloidin-Atto-488 and all other chemicals of research grade were obtained from Sigma-Aldrich Inc. (St Louis, MO, USA). All components for the magnetic separation of CD14 + monocytes were supplied by Miltenyi Biotec (San Diego, CA, USA). Human RANKL was procured from Insight Biotechnology (Middlesex, UK). Mouse RANKL and human M-CSF were acquired from R&D Systems (Minneapolis, MN). Alamar blue reagent, cell extraction buffer and NBT/BCIP Western Detection Chromogenic Kit were provided by Life Technologies (Carlsbad, CA, USA). Osteoassay surface multiwell plates were acquired from Corning Inc. (New York, NY, USA). The bicinchoninic acid (BCA) protein assay kit was purchased from Thermo Scientific (Rockford, IL, USA). Primary rabbit antibodies against total and phosphorylated p38, JNK and ERK were obtained from Signalway Antibody LLC (College Park, USA). Anti-GAPDH primary rabbit antibody was obtained from Abcam (Cambridge, MA, USA) and goat anti-rabbit ALP-conjugated secondary antibody was procured from Life Technologies (Carlsbad, CA, USA). pNiFty2-SEAP plasmids and Zeocin were purchased from InvivoGen (San Diego, CA, USA). BioCellin™ transfection reagent was obtained from BioCellChallenge (Cedex, France).
Cell culture of RAW264.7 murine macrophages
RAW264.7 murine macrophages (#TIB-71), were purchased from American Type Culture Collection (ATCC, Rockville, MD) and maintained in DMEM with 10 % FBS and antibiotic–antimycotic solution containing 100 U/ml penicillin, 100 μg/ml streptomycin and 0.25 μg/ml fungizone. Cells were incubated at 37 °C in a humidified atmosphere with 5 % CO2.
Isolation of human CD14+ monocytes and cell culture
All the procedures and experimental protocols were approved by the Human Research Ethics Committee of the Faculty of Health Sciences, University of Pretoria (Protocol approval number: 67/2014) and in accordance with the 1964 Helsinki declaration and its later amendments. Eligible participants were asked to provide an additional written informed consent for enrolment. Human CD14+ monocytes were isolated from peripheral blood (40–60 ml) of healthy male donors (aged 18–35) as described elsewhere (Kasonga et al. 2015), using CD14+ magnetic beads as per manufacturer’s instructions (Miltenyi Biotec, San Diego, CA, USA). Cells were cultured in α-MEM supplemented with 10 % FBS and antibiotic–antimycotic solution and were grown at 37 °C in a humidified atmosphere with 7 % CO2.
Stock solution
A 50 mM stock solution of EA was prepared in DMSO (vehicle) and frozen as aliquots in −80 °C until further use. Stock solutions were freshly diluted to working concentrations in complete culture medium before experiments. The final DMSO concentration in the culture medium did not exceed 0.1 % (v/v).
Alamar blue assay
Cells were seeded in 96-well plates and allowed to adhere for 12 h followed by exposure to increasing concentrations of EA (0.001, 0.01, 0.1, 1 µM) for 48 h. Alamar blue assay was conducted as per manufacturer’s instructions (Life Technologies). Absorbance was measured at 570 nm with 600 nm as reference wavelength on a microplate reader (BioTek Instruments Inc., Winooski, VT, USA).
Osteoclast differentiation
RANKL-treated RAW264.7 macrophages were differentiated into osteoclasts for 5 days as described previously (Deepak et al. 2015a). In brief, 5 × 103 cells per well were suspended in DMEM containing 10 % FBS and seeded into sterile 96-well culture plates. Cells were stimulated with RANKL alone (15 ng/ml) or in combination with increasing concentrations of EA. Cell culture media and factors were replaced every third day and differentiation was terminated on the fifth day unless otherwise stated.
CD14+ monocytes (4 × 104/well) were differentiated in the presence of M-CSF (25 ng/ml) and RANKL (30 ng/ml) for 14 days as described previously (Kasonga et al. 2015). Osteoclast-specific TRAP staining was assessed using a leukocyte acid-phosphatase kit as per manufacturer’s directions (Sigma-Aldrich, St Louis). TRAP+ cells with 3 or more nuclei were scored as osteoclasts. Photomicrographs were taken with a Zeiss Axiocam MRc5 camera attached to a Zeiss Axiovert 40 CFL microscope (Carl Zeiss AG, Oberkochen, Germany). The area covered by multi-nucleated osteoclasts/well was calculated by ImageJ software (Schneider et al. 2012).
Pit formation assay
The bone resorption activity of RANKL-induced osteoclasts derived from RAW264.7 macrophages was assessed using 24-well Corning osteoassay plates as reported elsewhere (Deepak et al. 2015b). Resorption pits were observed under a light microscope and quantified by ImageJ software.
Actin ring formation assay
Actin rings of osteoclasts were detected by staining actin filaments with Atto-conjugated phalloidin as described elsewhere (Boeyens et al. 2014). Images were acquired using a fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany).
Western blot analysis
Cell lysates were prepared using cell extraction buffer (Life Technologies, Carlsbad), supplemented with protease and phosphatase inhibitors (Sigma-Aldrich, St Louis) and resolved on 12 % SDS-PAGE gels. Purified proteins were quantified using a BCA protein assay kit as per manufacturer’s directions (Sigma-Aldrich, St Louis). Proteins were electrotransferred onto nitrocellulose membranes with Tris–glycine transfer buffer [25 mM Tris, 192 mM glycine, 20 % methanol (v/v)] probed with each antibody and detected by NBT/BCIP substrate. Digital images of the blots were acquired using a flatbed scanner (Ricoh Aficio, Johannesburg, South Africa).
NF-kB Secreted Embryonic Alkaline Phosphatase: Promoter (SEAP) assay
pNiFty2-SEAP (Invivogen) is an NF-kB-inducible reporter plasmid containing 5× NF-kB repeated transcription factor binding sites and a reporter gene—SEAP. RAW264.7 macrophages were stably transfected with GeneCellin™ transfection reagent. Stably transfected clones containing SEAP plasmid were selected with Zeocin. For promoter assay, the transfected cell line was stimulated with 35 ng of RANKL in the presence or absence of EA and SEAP assay was conducted after 48 h as per manufacturer’s protocol.
Mature-osteoclast survival assay
Osteoclasts were generated by RANKL treatment from RAW264.7 macrophages. Mature osteoclasts were exposed to EA for 48 h. At the end of treatment, cells were stained for TRAP and images were acquired using a Zeiss Axiocam MRc5 camera attached to a Zeiss Axiovert 40 CFL microscope (Carl Zeiss AG, Oberkochen, Germany).
qRT-PCR
RAW264.7 cells were seeded in 24-well plates at a density of 3 × 104/well with RANKL (15 ng/ml) or vehicle in the presence of EA for 5 days. Total RNA was isolated with TRI-reagent (Sigma) and 1 µg of the RNA was reverse transcribed into cDNA with MuMLV reverse transcriptase (New England Biolabs, UK) according to the manufacturer’s instructions. Resultant cDNA template was further utilized for conducting the qRT-PCR assay with gene specific primers for cathepsin-k (CTSK), dendritic cell-specific transmembrane protein (DC-STAMP), matrix metalloproteinase-9 (MMP-9) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (primer details available on request). GAPDH served as a loading control. PCR products were amplified using KAPA SYBR® FAST qPCR Kit Master Mix on a Roche LightCycler® Nano Instrument. Results were analyzed by the 2−∆∆C T method.
Statistical analysis
Data are representative of three independent experiments unless otherwise stated and are represented as mean ± standard deviation (SD). Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by Tukey post hoc multiple comparison test using Graph Pad Prism Software (GraphPad Software Inc., CA, USA). P < 0.05 was regarded as statistically significant.
Results
Effects of EA on cell viability
RAW264.7 macrophages and human CD14+ monocytes were treated with increasing concentrations (0.001, 0.01, 0.1, 1 µM) of EA (Fig. 1a) for 48 h. Cell viability was analyzed by alamar blue assay. At these concentrations EA did not have any cytotoxic effects (Fig. 1b, c).
EA inhibits osteoclast differentiation at an early stage of differentiation
We first examined the effects of EA on RANKL-induced osteoclast differentiation in RAW264.7 macrophages and human CD14+ monocytes by TRAP staining. EA dose-dependently inhibited RANKL-induced osteoclast formation in both the in vitro and ex vivo models. EA at a concentration as low as 0.1 µM had a significant effect on osteoclast formation and a peak inhibition was seen at 1 µM (Fig. 2a–d).

Effects of EA on RANKL-induced osteoclast formation. a, b RAW264.7 macrophages or c, d CD14+ monocytes were treated with RANKL in the presence or absence of EA as mentioned in materials and methods for 5 days and 14 days respectively (ns non-significant). e RAW264.7 macrophages were differentiated into osteoclasts in the presence of RANKL and the resultant mature osteoclasts were exposed to EA (1 µM) for 48 h. TRAP-positive cells containing more than three nuclei were scored as osteoclasts. Osteoclasts stain purple/red in the presence of TRAP (scale bars 20 µm). The results are mean ± SD and are representative of three independent experiments. (***P < 0.001 versus RANKL control)
Next, we studied whether EA could affect osteoclast numbers at later stages of differentiation. Mature osteoclasts derived from RAW264.7 macrophages at the 4th day of differentiation were treated with EA. Interestingly, EA did not affect mature osteoclast morphology or numbers (Fig. 2e, osteoclast quantification data not shown). Hence, EA inhibits osteoclastogenesis at an early stage of differentiation.
EA inhibits actin ring formation, bone resorptive function of osteoclasts and expression of osteoclast-specific markers
During differentiation, the mononuclear precursors fuse into multinucleated osteoclasts, a process that involves cytoskeleton rearrangement and actin ring formation. Actin rings aid osteoclasts in attachment to bone surface and resorption. Since EA had significant anti osteoclastogenic effects, we next sought to find out whether this effect correlated with disrupted actin ring formation and bone resorption. As seen in Fig. 3a, EA impaired fusion of mononuclear progenitors, resulting in disruption of actin ring formation which also led to a reduction in bone resorptive function of osteoclasts on bone-mimetic osteoassay plates (Fig. 3b). CTSK, DC-STAMP and MMP-9 are key osteoclast-specific genes induced by RANKL during osteoclastogenesis. We examined effects of EA on the mRNA expression of these genes. We found that EA downregulated the expression of the examined osteoclast-specific marker genes (Fig. 4).

Effects of EA on actin ring formation and bone resorption. a RAW264.7 macrophages were treated with RANKL in the presence or absence of EA as mentioned in materials and methods for 5 days. Differentiated osteoclasts were stained with phalloidin for actin ring formation and Hoechst was used as a nuclear stain (scale bars 20 µm). b RAW264.7 macrophages were seeded on osteoassay multi-well plates and assayed for resorption pit formation with RANKL in the presence or absence of EA for 7 days (scale bars 50 µm). Light areas are the resorbed surfaces. Resorption percentages are indicated in the figures

Effects of EA on osteoclast-specific gene expression. Analysis of osteoclast-specific genes was performed by qRT-PCR in RANKL-induced osteoclasts. The results are representative of three independent experiments. (*P < 0.05, ***P < 0.001 versus RANKL control)
EA suppresses RANKL-induced p38 MAP kinase pathway
RANKL-induced osteoclast formation involves activation of NF-kB and MAP kinase pathways. To elucidate the molecular mechanisms underlying EA-mediated inhibitory effects on osteoclastogenesis, we examined these pathways by SEAP- NF-kB promoter assay and western blot analysis. Figure 5a demonstrates that EA had no effect on NF-kB whereas it remarkably inhibited the activation and phosphorylation of the p38 MAP kinase pathway (Fig. 5b). However, activation of the ERK or JNK pathways remained unaffected suggesting that EA-mediated inhibition of osteoclast formation is due to the attenuation of the p38 pathway.

Effects of EA on RANKL signaling pathways. a NF-kB promoter activity was analyzed by SEAP assay as described in materials and methods with RANKL alone or in combination with EA (ns non-significant). b Cells were treated with RANKL alone or in combination with EA for 30 min. Post-incubation cell lysates were analyzed for the effects of EA on RANKL-induced MAPK pathways by western blot. The results are mean ± SD and are representative of three independent experiments. (**P < 0.01 versus vehicle control, ns = RANKL versus RANKL + EA)
Discussion
Bone loss diseases such as osteoporosis, cancer metastases to bone, rheumatoid arthritis and periodontitis involving alveolar bone loss are characterized by excessive osteoclast activity (Helfrich 2003). Hence, targeting osteoclast formation and activity is a promising approach for alleviating bone loss. We studied the effects of EA on osteoclast formation in murine RAW264.7 macrophages in vitro and human CD14+ monocytes ex vivo. Our results demonstrate that EA potently attenuates RANKL-induced osteoclastogenesis in osteoclast progenitor cells without cytotoxicity via suppression of the p38 signaling pathway.
Osteoclasts are multinucleated cells arising from the hematopoietic cells of monocyte/macrophage lineage (Novack and Teitelbaum 2008). Mature and functional osteoclasts express osteoclast-specific markers such as DC-STAMP, MMP-9 and CTSK. DC-STAMP is pivotal for the fusion of precursor cells (Yagi et al. 2005). Proteases such as CTSK and MMP-9 aid in bone matrix resorption (Inui et al. 1999). CTSK is an osteoclast-specific enzyme and drugs targeting CTSK are currently in clinical trials (Bone et al. 2010). Mice lacking CTSK have osteoclasts with impaired bone resorptive potential and suffer from osteopetrosis (Gowen et al. 1999). Mutation in the CTSK gene has been reported to cause pycnodysostosis (a disorder with dense bones) (Gelb et al. 1996). MMP-9 is expressed by active osteoclasts and plays an important role in osteoclast-mediated bone resorption. MMP-9 knockout mice exhibit a delay in osteoclast recruitment during early bone development (Vu et al. 1998). In this study we found that EA potently inhibited the expression of DC-STAMP, CTSK and MMP-9 during osteoclast differentiation.
NF-kB and MAP kinase signaling pathways (JNK, ERK and p38) play an important role in RANKL-induced osteoclast formation (Wada et al. 2006; Soysa and Alles 2009). Genetic studies have shown that the NF-kB pathway is crucial for normal skeletal development and bone health. Disruption of this pathway results in impaired osteoclast differentiation and/or function and abnormal skeletal development (Otero et al. 2012). Abrogation of the ERK pathway has been reported to result in reduced osteoclast progenitor cell numbers and decreased osteoclast formation (He et al. 2011). Using mice devoid of JNK or expressing a dominant-negative form of JNK, researchers have shown that the JNK pathway is indispensable for efficient osteoclastogenesis (Ikeda et al. 2008). The p38 pathway activation has been shown to be vital during early stages of RANKL-induced osteoclast differentiation (Li et al. 2002). By pharmacological inhibition, expression of the dominant-negative form of p38 or genetic ablation of p38, researchers have shown that this MAP kinase is essential for osteoclast differentiation. Mice lacking p38 are deficient in osteoclasts (Matsumoto et al. 2000; Li et al. 2002). We analyzed the effects of EA on the NF-kB and MAPKs signaling pathways and found that EA inhibited the phosphorylation of p38 without affecting the NF-kB or ERK/JNK pathways. These results indicated that the inhibitory effects of EA on activation of the p38 pathway may contribute to its anti-osteoclastogenic effects. Furthermore, we found that EA-mediated suppressive effects on osteoclastogenesis were confined only to the early stage of differentiation, as we did not observe a change in osteoclast numbers when the mature multinucleated cells were exposed to EA. The NF-kB and ERK/JNK pathways have been shown to play an important role in osteoclast survival and exert anti-apoptotic effects. Since EA had no effect on the NF-kB and ERK/JNK pathways but only affected the p38 MAPK pathway, it could explain its inefficiency to affect mature osteoclast survival.
Actin ring formation is a characteristic hallmark feature of osteoclasts. Actin rings aid osteoclasts in bone resorption via formation of a sealing zone within the resorption lacunae and attachment to the extracellular bone matrix (Teitelbaum 2000). Our results demonstrate that EA, by attenuating fusion of osteoclasts, not only disrupted actin ring formation but severely affected the bone resorptive function of osteoclasts.
Taken together, this study demonstrates that EA inhibits RANKL-induced osteoclast differentiation in murine RAW264.7 macrophages in vitro and human CD14+ monocytes ex vivo. EA suppressed the expression of osteoclastic specific marker genes, disrupted actin ring formation and aggravated bone resorption. Elucidation of the underlying molecular mechanisms revealed that EA attenuated RANKL-induced activation of the p38 MAP kinase pathway. Hence, EA is a novel anti-osteoclastogenic molecule and should be explored further for its potential as a therapeutic option for diseases characterized by excessive osteoclast activity.
References
Augustine M, Horwitz MJ (2013) Parathyroid hormone and parathyroid hormone-related protein analogs as therapies for osteoporosis. Curr Osteoporos Rep 11:400–406
Boeyens JC, Deepak V, Chua WH, Kruger MC, Joubert AM, Coetzee M (2014) Effects of omega3- and omega6-polyunsaturated fatty acids on RANKL-induced osteoclast differentiation of RAW264.7 cells: a comparative in vitro study. Nutrients 6:2584–2601
Bone HG, Mcclung MR, Roux C, Recker RR, Eisman JA, Verbruggen N, Hustad CM, Dasilva C, Santora AC, Ince BA (2010) Odanacatib, a cathepsin-K inhibitor for osteoporosis: a two-year study in postmenopausal women with low bone density. J Bone Miner Res 25:937–947
Boyce BF, Xing L (2008) Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys 473:139–146
Charles JF, Aliprantis AO (2014) Osteoclasts: more than ‘bone eaters’. Trends Mol Med 20:449–459
Chen JS, Sambrook PN (2012) Antiresorptive therapies for osteoporosis: a clinical overview. Nat Rev Endocrinol 8:81–91
Deepak V, Kasonga A, Kruger MC, Coetzee M (2015a) Inhibitory effects of eugenol on RANKL-induced osteoclast formation via attenuation of NF-kappaB and MAPK pathways. Connect Tissue Res 56:195–203
Deepak V, Kruger MC, Joubert A, Coetzee M (2015b) Piperine alleviates osteoclast formation through the p38/c-Fos/NFATc1 signaling axis. BioFactors 41:403–413
Gelb BD, Shi GP, Chapman HA, Desnick RJ (1996) Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 273:1236–1238
Gowen M, Lazner F, Dodds R, Kapadia R, Feild J, Tavaria M, Bertoncello I, Drake F, Zavarselk S, Tellis I, Hertzog P, Debouck C, Kola I (1999) Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res 14:1654–1663
He Y, Staser K, Rhodes SD, Liu Y, Wu X, Park SJ, Yuan J, Yang X, Li X, Jiang L, Chen S, Yang FC (2011) Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS One 6:e24780
Helfrich MH (2003) Osteoclast diseases. Microsc Res Tech 61:514–532
Helfrich MH (2005) Osteoclast diseases and dental abnormalities. Arch Oral Biol 50:115–122
Hofbauer LC, Heufelder AE, Erben RG (2001) Osteoprotegerin, RANK, and RANK ligand: the good, the bad, and the ugly in rheumatoid arthritis. J Rheumatol 28:685–687
Ikeda F, Matsubara T, Tsurukai T, Hata K, Nishimura R, Yoneda T (2008) JNK/c-Jun signaling mediates an anti-apoptotic effect of RANKL in osteoclasts. J Bone Miner Res 23:907–914
Inui T, Ishibashi O, Origane Y, Fujimori K, Kokubo T, Nakajima M (1999) Matrix metalloproteinases and lysosomal cysteine proteases in osteoclasts contribute to bone resorption through distinct modes of action. Biochem Biophys Res Commun 258:173–178
Kasonga AE, Deepak V, Kruger MC, Coetzee M (2015) Arachidonic acid and docosahexaenoic acid suppress osteoclast formation and activity in human CD14+ monocytes, in vitro. PLoS One 10:e0125145
Li X, Udagawa N, Itoh K, Suda K, Murase Y, Nishihara T, Suda T, Takahashi N (2002) p38 MAPK-mediated signals are required for inducing osteoclast differentiation but not for osteoclast function. Endocrinology 143:3105–3113
Marquardt KC, Watson RR (2014) Polyphenols and Public Health. In: Watson RR, Preedy VR, Zibadi S (eds) Polyphenols in Human Health and Disease, 1st edn. Academic Press, San Diego, pp 9–15
Matsumoto M, Sudo T, Maruyama M, Osada H, Tsujimoto M (2000) Activation of p38 mitogen-activated protein kinase is crucial in osteoclastogenesis induced by tumor necrosis factor. FEBS Lett 486:23–28
Novack DV, Teitelbaum SL (2008) The osteoclast: friend or foe? Annu Rev Pathol 3:457–484
Otero JE, Chen T, Zhang K, Abu-Amer Y (2012) Constitutively active canonical NF-kappaB pathway induces severe bone loss in mice. PLoS One 7:e38694
Rachner TD, Khosla S, Hofbauer LC (2011) Osteoporosis: now and the future. Lancet 377:1276–1287
Rodan GA, Martin TJ (2000) Therapeutic approaches to bone diseases. Science 289:1508–1514
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675
Shen CL, Von Bergen V, Chyu MC, Jenkins MR, Mo H, Chen CH, Kwun IS (2012) Fruits and dietary phytochemicals in bone protection. Nutr Res 32:897–910
Soysa NS, Alles N (2009) NF-kappaB functions in osteoclasts. Biochem Biophys Res Commun 378:1–5
Teitelbaum SL (2000) Bone resorption by osteoclasts. Science 289:1504–1508
Usta C, Ozdemir S, Schiariti M, Puddu PE (2013) The pharmacological use of ellagic acid-rich pomegranate fruit. Int J Food Sci Nutr 64:907–913
Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z (1998) MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93:411–422
Wada T, Nakashima T, Hiroshi N, Penninger JM (2006) RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med 12:17–25
Walsh MC, Choi Y (2014) Biology of the RANKL-RANK-OPG System in immunity, bone, and beyond. Front Immunol 5:511
Whitaker M, Guo J, Kehoe T, Benson G (2012) Bisphosphonates for osteoporosis–where do we go from here? N Engl J Med 366:2048–2051
Yagi M, Miyamoto T, Sawatani Y, Iwamoto K, Hosogane N, Fujita N, Morita K, Ninomiya K, Suzuki T, Miyamoto K, Oike Y, Takeya M, Toyama Y, Suda T (2005) DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med 202:345–351
Acknowledgments
This study was supported by Grants from RESCOM, University of Pretoria; the University of Pretoria’s Strategic Institutional Research Theme in Food, Nutrition and Well-being; and in part by the University of Pretoria Vice Chancellor’s Postdoctoral Research Fellowship.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Rights and permissions
About this article

Cite this article
Rantlha, M., Sagar, T., Kruger, M.C. et al. Ellagic acid inhibits RANKL-induced osteoclast differentiation by suppressing the p38 MAP kinase pathway. Arch. Pharm. Res. 40, 79–87 (2017). https://doi.org/10.1007/s12272-016-0790-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0790-0