
Abstract
Timosaponin AIII (AIII), a steroidal saponin isolated from Anemarrhena asphodeloides Bge. Our study showed that AIII induced both apoptosis and autophagy, and autophagy inhibited apoptosis in A375S2 cells. Furtherly, this study was carried out to investigate what kind of cytokines plays an important role in this process. The results revealed that AIII induced apoptosis through activating c-Jun N-terminal protein kinase (JNK) or extracellular signal related kinase (ERK) signaling pathway and generating NO. However, JNK or ERK inhibited autophagy, while NO had no effect on autophagy. Therefore, JNK, ERK or NO regulates two programmed death processes in different ways. AIII did not show obvious cytotoxic effect on human peripheral blood mononuclear cells, which indicated that AIII has less side effects on normal cells, and could be considered as a leading compound for developing novel anticancer drug.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Malignant melanoma is a devastating skin cancer. In recent years, the incidence of malignant melanoma has increased. As reported, the RAS/RAF/MEK/ERK signaling pathway is important for the pathogenesis of malignant melanoma. Mutations in this signaling pathway occur in 50–70 % malignant melanoma (Wang and Qi 2013) and activated ERK MAPK was found in 90 % of human melanomas (Qin et al. 2012). The c-Jun N-terminal protein kinase (JNK), the extracellular signal related kinase (ERK) and the p38 protein kinase are members of mitogen-activated protein kinases (MAPKs), which can be activated by distinct mitogens play an essential role in cell growth, differentiation, apoptosis and autophagy (Liu et al. 2012). In this study, we examined the effect of MAPKs on the induction of apoptosis and autophagy in AIII- treated A375-S2 cells.
NO is an important reactive free radical synthesized by nitric oxide synthases (NOS). Three isoforms known as NOS: endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS). Three NOSs have the specific expressions in a variety of cancer cell types. Under different conditions NO has dual actions against tumour, depending on its concentration, time or sites. Low concentrations of NO may promote cancer cell growth or metastasis, but high concentrations of NO may induce cancer cell death through apoptosis pathway (Zhang et al. 2005). NO is thought to play an important role in the fate of A375-S2 cells treated with AIII.
Timosaponin AIII shows pharmacological activities such as anti-aging, anti-depressant, anti-diabetic etc. (Bao et al. 2007). Its antitumor activity has been described for different types of cancer. AIII was reported to inhibit the cytotoxic and anti-neoplastic activity in human colon cancer HCT-15 cells (Kang et al. 2011). AIII induced autophagy, preceding mitochondria-mediated apoptosis, in human cervical cancer HeLa cells (Sy et al. 2008). Furthermore, it also suppressed the invasive activity induced by hepatocyte growth factor through sustained ERK activation in human breast cancer MDA-MB-231 cells (Tsai et al. 2013). Reports on simultaneous induction of apoptosis and autophagy with AIII are not well documented. The effects of AIII on malignant melanoma cells remain unclear. Compared with other melanoma cells, A375-S2 cells have the higher expression of insulin-like growth factor 1 (IGF-1), a major survival factor (Kanter-Lewensohn et al. 2000). Therefore we focused on the A375-S2 cells as the target to test the anti-tumor effect of AIII.
Materials and methods
Chemicals and reagents
Timosaponin AIII was isolated from Anemarrhena asphodeloides Bge. and its purity was 95.0 %. AIII was dissolved in dimethyl sulfoxide (DMSO) as a stock solution. The DMSO concentration was kept below 0.1 % so that DMSO did not show any detectable effect on cell viability. 5-FU and paclitaxel were obtained from Institution of Pharmaceutical and Biological Products Chinese. The chromatographic silica gel (200–300 mesh) was obtained from Qingdao Ocean Chemical Factory, and Macroporous adsorption resin D101 was produced by Tianjin Rubber Factory. Fetal bovine serum (FBS) was obtained from TBD Biotechnology Development; MEM was obtained from Grand Island Biotechnology. Ficoll–Hypaque solution (density 1.077 g/ml) was purchased from Tianjin Chuanye Biological Products. 4,5-diaminofluo-rescein diacetate (DAF-2DA), acridine orange (AO), 3-methyladenine (3-MA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), propidium iodide (PI), monodansylcadaverine (MDC) and anti-LC3 antibody were purchased from Sigma Chemical. JNK MAPK inhibitor SP600125, ERK MAPK inhibitor PD98059, iNOS inhibitor N-(3-(aminomethyl) benzyl) acetamidine (1400 W) and NO scavenger dithiothreitol (DTT) were purchased from Calbiochem. Anti-caspase-3, iNOS, Beclin-1, JNK, ERK, P38, β-actin antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from Santa Cruz Biotechnology. Electrochemiluminescence (ECL) reagent was from Thermo Scientific.
The extraction and isolation of AIII
The rhizomes of A. asphodeloides Bge. were collected from Shijiazhuang, Hebei province, People’s Republic of China, in October 2011, and were identified by Professor Lu J. C. (Department of Natural Products Chemistry, Shenyang Pharmaceutical University, People’s Republic of China). A voucher specimen (No. 20120403) has been deposited in the Herbarium of Shenyang Pharmaceutical University, Liaoning, People’s Republic of China.
The air-dried rhizomes of A. asphodeloides Bge. (10.0 kg) were crushed to pieces and extracted in H2O for 3 × 30 l × 5 h at 80–90 °C. The combined extracts were concentrated to give a crude extract (860 g), which was suspended in H2O and partitioned with n-BuOH. The n-BuOH extract (490 g) was suspended in H2O and then chromatographed over D101 macroporous resin column using H2O–EtOH (100:0, 30:70 and 5:95) as eluents. The H2O–EtOH (30:70) fraction (206 g) was subjected to silica gel CC eluted with CHCl3–MeOH (from 100:1 to 1:1) to afford thirty fractions (1–30). Further purification of fraction 4 by recrystallizing from EtOH yielded timosaponin AIII (3.2 g).
Cell culture
A375-S2 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). They were cultured in MEM medium supplemented with 10 % foetal bovine serum (FBS), 100 μg/ml streptomycin and 100 U/ml penicillin. The cells were incubated with 5 % CO2 in a humidified atmosphere at 37 °C. The cells in the exponential phase of growth were used in the experiments.
Preparation of human peripheral blood mononuclear cells (hPBMC) (Bigaud et al. 2004; Nilsson et al. 2008)
Fresh peripheral blood was collected from 3 healthy human donors (males) in tubes containing 3.2 % sodium citrate. The peripheral blood was diluted 1:3 in PBS and transferred 30 ml to each centrifuge tubes in an ice bath environment. Then 10 ml of Ficoll–Hypaque solution was slowly pipetted into the bottom of centrifuge tubes and centrifuged at 900×g, 18–20 °C for 25 min. Then the mononuclear cells were pipetted into new centrifuge tubes and washed in PBS at 400×g, 18–20 °C for 10 min for three times and resuspended in RPMI-1640 medium containing 2 % heat inactivated human AB serum before counting. Cells were incubated in 96-well cell culture clusters (Corning, NY, USA) at a density of 5 × 104 cells per well for 3 h. Then the hPBMC were treated with 5-FU, paclitaxel as positive control and AIII at 2.5–40 μM for 24 h, and the cell viability was measured by the MTT assay.
Growth inhibition assay
The inhibitory ratio was measured by the MTT assay. A375-S2 cells were incubated in 96-well cell culture plates at a density of 6 × 103 cells per well for 24 h. Then the cells were treated with different doses of AIII or treated with 4.5 μM AIII in the presence or absence of 3-MA and different concentrations of SP600125 (SP) or PD98059 (PD) for 24 h. After that, the cells were rinsed with ice-cold PBS once and incubated with 100 μl 0.5 mg/ml MTT at 37 °C for 2 h. Then the formazan crystals were dissolved with 150 μl DMSO and the absorbance were measured at 492 nm by a microplate reader (Thermo Scientific Multiskan MK3, Shanghai, China). The inhibitory ratio of cells was calculated as follows:
Observation of morphological changes
A375-S2 cells were incubated in 96-well cell culture plates at a density of 6 × 103 cells per well for 24 h. Then the cells were treated with AIII for another 24 h. The cell morphology was observed with a phase contrast microscope (Leica, Nussloch, Germany).
Observation of apoptosis and autophagy by fluorescence microscopy
A375-S2 cells were incubated in 24-well cell culture plates at a density of 6 × 104 cells per well for 24 h. Then the cells were treated with different concentrations of AIII for another 24 h. For observing apoptosis, the cells were stained with 20 μg/ml AO and immediately examined by a fluorescence microscope (Olympus, Tokyo, Japan). For detecting autophagy, the cells were stained with 0.5 mg/ml MDC at 37 °C for 30 min, then rinsed with ice-cold PBS once before observing by the fluorescence microscope.
Analysis of apoptosis by FACScan flow cytometry
A375-S2 cells were incubated in 6-well cell culture clusters at a density of 18 × 104 cells per well for 24 h. Then the cells were treated with different concentrations of AIII or with 4.5 μM AIII in the presence or absence of 3-MA for another 24 h. After harvesting and rinsing by PBS, the cells were fixed with 500 μl PBS and 10 ml 70 % ice-cold ethanol at 4 °C for at least 18 h. After that, the cells were washed by PBS twice and resuspended with 500 μl PI solution (PI 50 μg/ml and RNase A 1 mg/ml) in an ice bath environment for 1 h. Then the cell apoptosis was detected by FACScan flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA).
Analysis of NO by FACScan flow cytometry
A375-S2 cells were incubated in 6-well cell culture plates at a density of 18 × 104 cells per well for 24 h. Then the cells were treated with different concentrations of AIII or treated with 4.5 μM AIII for 24 h in the presence of 1 mM DTT or 120 μM 1400 W. After harvesting and rinsing with PBS, the cells were resuspended with 10 μM NO specific dye DAF-2DA and incubated at 37 °C for 30 min. The samples were analyzed by FACScan flow cytometry.
Western blot analysis
A375-S2 cells were incubated in 25 ml culture bottle at a density of 3 × 105 per bottle, then treated with different concentrations of AIII or inhibitors for another 24 h. After harvesting the supernatant and washing cells by PBS twice, the cells were lysed in whole cell RIPA lysis buffer (Beyotime, Haimen, Jiangsu, China) supplemented with PMSF (phenylmethanesulfonyl fluoride) (1 mM) at 4 °C for 1 h. After 15,000×g centrifugation at 4 °C for 10 min, the protein content of the supernatant was determined by the Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA, USA). The same amounts of the total protein were separated by 12 % SDS-polyacrylamide gel electrophoresis and transferred to Millipore Immobilon-P Transfer Membrane (Millipore, Billerica, MA, USA). The membranes were soaked in 5 % skimmed milk and the proteins were incubated with the primary antibodies overnight, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies and detected by using ECL as the HRP substrate. For all of Western blot experiments, the authors have carried out for several times, and show the representation of results.
Statistical analysis
All the data from at least three independent experiments were expressed as mean ± SD. Statistical comparisons were made by one-way ANOVA followed by Statistics Package for Social Science software (version 13.0; SPSS, Chicago, IL, USA). p < 0.05 were considered to be a statistically significant difference.
Results
AIII had no significant cytotoxicity toward hPBMC but inhibited A375-S2 cell growth
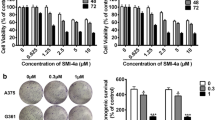
We isolated hPBMC from three healthy male volunteers and then the hPBMC were treated with AIII at 2.5–40 μM for 24 h compared with the same concentrations of 5-fluorouracil (5-FU) and paclitaxel. The cell viability of hPBMC treated with AIII was similar to those treated with 5-FU and paclitaxel at lower concentrations (2.5–20 μM). At a high concentration (40 μM), the cell viability appeared to be lower than those treated with 5-FU and paclitaxel with significant differences. However, the drug concentration (4.5 μM) is much lower than the high concentration (40 μM) in our experiments. These results showed AIII had no obvious cytotoxic effect on hPBMC by MTT assay (Fig. 1b). By contrast, AIII induced the growth inhibition of A375-S2 cells in a concentration-dependent manner at 24 h (Fig. 1c).

The growth inhibitory activity of AIII in normal human cells (hPBMC) and A375-S2 cells. a The chemical structure of AIII. b AIII-induced growth inhibition in normal human cells (hPBMC) compared with 5-fluorouracil (5-FU) and paclitaxel was determined by MTT assay. The hPBMC were treated with AIII, 5-FU and paclitaxel at 2.5–40 μM for 24 h. c A375-S2 cells were treated with various concentrations of AIII for 24 h. The inhibitory ratio was measured by the MTT assay (n = 3, mean ± SD)
AIII induced apoptosis in A375-S2 cells
A375-S2 cells seeded either in 24 or 6-well, were treated with 4.5 μM AIII for another 24 h. The cell morphology was observed by phase contrast microscopy. Smaller cells and granular apoptotic bodies were observed. 20 μg/ml AO staining showed significant chromatin condensation with dense green fluorescence compared with control (Fig. 2a). After the cells were incubated with different concentrations of AIII for 24 h followed by staining with PI, cell cycle stages were measured by flow cytometric analysis. AIII induced increased SubG1 fraction in a concentration-dependent manner (Fig. 2b). Cell apoptosis is one type of the programmed cell death (PCD) and cleaved caspase-3 activity is the marker. Western blot analysis revealed that the protein levels of cleaved caspase-3 increased in a dose-dependent manner (Fig. 2c). These results showed the onset of apoptosis.

AIII induced apoptosis in A375-S2 cells. a A375-S2 cells were incubated with 4.5 μM AIII for 24 h, and the morphological changes were observed by phase-contrast microscopy (×200 magnification) or by fluorescence microscopy with AO staining (×200 magnification). b and c The cells were incubated with different concentrations of AIII for 24 h. b After staining with PI, the cells were analysed by flow cytometry, and the subG1 fraction calculated is shown on the lower panel (n = 3, mean ± SD), c The protein levels of caspase-3 were examined by Western blot analysis
AIII induced autophagy in A375-S2 cells
MDC is a specific dye for autophagy vacuole. Its fluorescent intensity reflects autophagic ratio. A375-S2 cells were incubated with 4.5 μM AIII for 24 h and stained with MDC at 37 °C for 1 h, then the changes of cell morphology were observed by fluorescence microscopy. Compared with control, the cells treated with 4.5 μM AIII had stronger fluorescent intensity of intracellular MDC (Fig. 3a). Western blot analysis revealed that the up-regulation of the conversion of LC3 I to LC3 II (a biological marker for autophagy vacuole formation) (Jiang et al. 2011) and the protein levels of Beclin-1 increased in AIII-treated cells in a dose-dependent manner. These results showed that AIII induced autophagy in A375-S2 cells (Fig. 3b).

AIII induced autophagy in A375-S2 cells. a A375-S2 cells were incubated with 4.5 μM AIII for 24 h, and the morphological changes were observed by fluorescence microscopy with MDC staining (×200 magnification). Arrows indicate cells containing autophagolysosome. b The cells were incubated with different concentrations of AIII for 24 h. The protein levels of LC3 and Beclin-1 were examined by Western blot analysis
Autophagy antagonized apoptosis in AIII-treated A375-S2 cells
To determine the relationship between apoptosis and autophagy in AIII-treated A375-S2 cells, 3-methyladenine (3-MA), a specific autophagic inhibitor, was utilized. We examined the effect of 3-MA on AIII-induced autophagy by the expression of LC3 I/II. A375-S2 cells were pre-treated with 1 mM 3-MA for 1 h, and then co-incubated with 4.5 μM AIII for another 24 h. As shown in Fig. 4a, the conversion from LC3 I to LC3 II were reversed by 3-MA in AIII-treated A375S2 cells. It suggests that 3-MA can inhibit AIII-induced autophagy in A375S2 cells. An MTT assay demonstrated that AIII inhibited cell growth significantly, but this inhibition was partially augmented by 3-MA (Fig. 4b). Subsequently, the expression of apoptosis-related proteins was detected by Western blot. Treatment with AIII elevated the cleavage of procaspase-3, whereas this enhancement was further up-regulated by 3-MA (Fig. 4c). Then, the apoptosis ratio was detected by flow cytometry, and it showed that 3-MA-AIII treatment increased the apoptotic ratio compared with AIII treatment alone (Fig. 4d). In short, 3-MA suppressed autophagy and increased the apoptotic ratio induced by AIII, indicating that autophagy can antagonize apoptosis in AIII-treated A375-S2 cells.

Autophagy antagonized apoptosis in AIII-treated A375-S2 cells a–d A375-S2 cells were incubated with 4.5 μM AIII in the presence or absence of 3-MA (1 mM) for 24 h. a Effects of 3-MA (1 mM) on the expression LC3 I/II detected by Western blot analysis. b The inhibitory ratio was measured by the MTT assay (n = 3, mean ± SD). c Protein levels of caspase-3 were examined by Western blot analysis. d PI positive cells were detected by flow cytometry (n = 3, mean ± SD)
AIII induced NO generation in A375-S2 cells; NO played an important role in apoptosis but not in autophagy
We used DAF-2DA (a type of NO fluorescent dyes) to detect NO generation. The results showed NO generation increased in a dose-independent manner by flow cytometry (Fig. 5a). Western blot analysis revealed that the expression of iNOS was coincided with the DAF-2T positive ratio (Fig. 5b). Since AIII induced NO generation in A375-S2 cells, we examined the role of NO in induction of apoptosis and autophagy. A375-S2 cells were pre-treated with 1 mM DTT or 120 μM 1400 W as iNOS inhibitors for 1 h, and then co-incubated with 4.5 μM AIII for another 24 h. Detected by flow cytometry, DTT and 1400 W reduced NO generation induced by AIII (Fig. 5c). Compared with the group treated with AIII alone, the protein levels of cleaved caspase-3 were markedly decreased in the presence of DTT and 1400 W (Fig. 5d). But no change was observed in the conversion of LC3 I to LC3 II in the presence of DTT and 1400 W (Fig. 5e). These results showed that NO induced the onset of apoptosis but did not contribute to autophagy induction.

AIII induced-NO generation, promoting the apoptosis in A375-S2 cells. a and b A375-S2 cells were treated with different concentrations of AIII for 24 h, a DAF-2T positive cells were detected by flow cytometry (n = 3, mean ± SD) or the expression of iNOS was examined by Western blot analysis. b, c–e The cells were treated with 4.5 μM AIII alone for 24 h or the cells were pre-treated with 1 mM DTT or 120 μM 1400 W for 1 h, respectively, followed by coincubation with 4.5 μM AIII for another 24 h. c DAF-2T positive cells were detected by flow cytometry. *p < 0.05 versus AIII alone-treated group. The protein levels of caspase-3 (d) and LC3 (e) were examined by Western blot analysis
JNK and ERK were involved in the induction of apoptosis and autophagy in AIII-treated A375-S2 cells
Mutations in the RAS/RAF/MEK/ERK signaling pathway occurred in 50–70 % malignant melanoma (Wang and Qi 2013). So we examined the effects of MAPKs pathways on the apoptosis and autophagy of AIII-treated A375-S2 cells. The cells were treated with different concentrations of AIII for 24 h. Western blot analysis revealed that the expression of both p-JNK and p-ERK increased. While the protein levels of p-P38 had no obvious change, P38 expression was decreased (Fig. 6a). We chose the effective concentrations of SP600125 (the JNK inhibitor) and PD98059 (the ERK inhibitor) by the MTT assay for pre-treatment of A375-S2 cells with 2 and 2.5 μM, respectively, for 1 h and then co-incubated with 4.5 μM AIII for another 24 h (Fig. 6b). Western blot analysis revealed that compared with the group treated with AIII alone, the protein levels of cleaved (active) caspase-3 markedly decreased, but the conversion of LC3 I to LC3 II increased in the presence of SP or PD. These results showed that JNK and ERK promoted the onset of apoptosis but inhibited autophagy in A375-S2 cells (Fig. 6c, d).

AIII increased the expression of JNK and ERK in A375-S2 cells, which promoted the apoptosis but inhibited autophagy in A375-S2 cells. a A375-S2 cells were treated with different concentrations of AIII for 24 h, and the expression of p-P38, p-JNK and p-ERK were examined by Western blot analysis. Data shown are representative of three separate experiments. b A375-S2 cells were treated with 4.5 μM AIII for 24 h in the presence or absence of different concentrations of SP600125 (SP) (b-1) or PD98059 (PD) (b-2). The inhibitory ratio was measured by the MTT assay. c and d A375-S2 cells were treated with 4.5 μM AIII for 24 h, in the presence or absence of 2 μM SP600125 (SP) or 2.5 μM PD98059 (PD), and the protein levels of caspase-3 (c) and LC3 (d) were examined by Western blot analysis
Discussion
Autophagy, the type II programmed cell death process, degraded cytoplasmic components on the double membrane vesicles by fusion with lysosomal compartments (Kundu and Thompson 2008). Autophagy sometimes occurs with apoptosis in the process of programmed cell death. It plays a critical role in removing damaged or surplus organelles in order to maintain cellular homeostasis (Yang et al. 2008a). Autophagy has complexed roles in cancer, which either promotes or inhibits tumorigenesis. The effects of autophagy depend on tumor type, stage, drug and others (Kimmelman 2011). Three types of interplay between autophagy and apoptosis have been reported: (1) autophagy and apoptosis both lead to cell death, (2) autophagy promotes cell survival by antagonizing apoptosis, and (3) autophagy leads to cell death by promoting apoptotic process (Eisenberg-Lerner et al. 2009). In the case of the steroidal saponin isolated from A. asphodeloides Bge, the previous studies showed apoptosis induction in human colon cancer HCT-15 cells, HepG2 human hepatoma cells and other types of cells. However, few focused on the induction of autophagy in the past. This research team undertook a preliminary study on antitumor activity of AIII, which were evaluated for their anti-tumor effects on ten tumor cell lines. Our study demonstrated that AIII induced both apoptosis and autophagy, which were related to NO, JNK or ERK in A375-S2 cells, and autophagy inhibited apoptosis in A375S2 cells. Autophagy may have an important role in the growth of A375-S2 cells. After understanding this mechanism, AIII in combination with autophagy inhibitors maybe play better anti-cancer effect in the clinical treatment of melanoma.
Reactive oxygen species (ROS) is a reactive free radical, including hydroxyl radical, superoxide anion, singlet oxygen and hydrogen peroxide. ROS have critical functions in cell proliferation, differentiation and apoptosis. In general, the high level of ROS for prolonged period causes damage to tumor cells, while the low level of ROS could activate pro-survival pathway (Fiers et al. 1999; Yu et al. 2012). In this study, AIII at low concentrations showed no significant effect on ROS generation, while AIII at 6 μM decreased the level of ROS slightly at 12 and 24 h. Since AIII did not induce obvious ROS generation, we tested on NO generation.
NO is another important reactive free radical synthesized by NOS. NO participates in many signaling pathways. Previous studies reported that NO induced apoptosis and autophagy. Autophagy in turn down-regulated NO synthesis in physalin A-treated A375-S2 cells (He et al. 2014). NO induced apoptosis and cell cycle arrest in evodiamine-treated A375-S2 cells (Yang et al. 2008b). This study indicated that NO induced apoptosis in AIII-treated A375-S2 cells, which was consistent with the previous studies. The results showed that high concentrations of NO induced melanoma cell death. But NO did not participate in the initiation of autophagy. Autophagy contributes to pro-survival or pro-death for different tumor cell types, depending on different conditions.
As reported, depending on cell lines and stimuli used, distinct MAPK subtypes were activated. Each MAPK subtype may have a different functional role (Suzuki et al. 2001). In general, the Ras/Raf/MEK/ERK cascade could be activated by the epidermal growth factors in relation with cell proliferation (Siegfried et al. 2013). Our speculation on the reason why ERK promoted the onset of apoptosis in this study is that ERK has the different isoforms: ERK1 and ERK2. The two isoforms may have different functions in melanoma cells. For example, reducing ERK1 but not ERK2 could block the mitochondrial and death receptor-related apoptosis pathways (Wang and Qi 2013). Besides, ERK activity can promote apoptotic pathways by influencing the mitochondrial function (Cagnol and Chambard 2010). In accordance with the apoptosis induction by ERK, ERK could be target for treating melanoma. In contrast with ERK, JNK activation is related to the initiation of apoptosis in response to cytokines, UV or cellular stress. In this study, JNK also induced the onset of apoptosis, while the protein levels of p-P38 did not show significant changes with the increasing of AIII concentration. In summary, AIII induced apoptosis through activating JNK or ERK signaling pathway and generating NO. JNK or ERK inhibited autophagy, while NO had no effect on autophagy. JNK, ERK or NO regulates two programmed death processes in different ways.
Malignant melanoma is one of the most serious skin cancers. In recent decades, the incidence has arisen quickly. About 48,000 people die from melanoma one year worldwide. Melanoma is highly resistant to conventional chemotherapy (Bay et al. 2015; Gibot et al. 2015). So it is urgent to develop a new drug to treat the melanoma. The findings that AIII induced apoptosis and autophagy in A375-S2 cells at lower concentration, indicating a possibility that AIII could inhibit tumor cell growth with less side effects from the results that the cytotoxicity of AIII on hPBMC was similarly lower to paclitaxel or 5-FU. Timosaponin AIII could be considered as a leading compound for developing novel anticancer drug.
References
Bao W, Pan H, Lu M, Ni Y, Zhang R, Gong X (2007) The apoptotic effect of sarsasapogenin from Anemarrhena asphodeloides on HepG2 human hepatoma cells. Cell Biol Int 31:887–892
Bay C, Kejs AM, Storm HH, Engholm G (2015) Incidence and survival in patients with cutaneous melanoma by morphology, anatomical site and TNM stage: a Danish population-based register study 1989–2011. Cancer Epidemiol 39:1–7
Bigaud M, Maurer C, Vedrine C, Puissant B, Blancher A (2004) A simple method to optimize peripheral blood mononuclear cell preparation from cynomolgus monkeys and improve mixed lymphocyte reactions. J Pharmacol Toxicol Methods 50:153–159
Cagnol S, Chambard JC (2010) ERK and cell death: mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. FEBS J 277:2–21
Eisenberg-Lerner A, Bialik S, Simon HU, Kimch A (2009) Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 16:966–975
Fiers W, Beyaert R, Declercq W, Vandenabeele P (1999) More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene 18:7719–7730
Gibot L, Chabaud S, Bouhout S, Bolduc S, Auger FA, Moulin VJ (2015) Anticancer properties of chitosan on human melanoma are cell line dependent. Int J Biol Macromol 72:370–379
He H, Feng YS, Zang LH, Liu WW, Ding LQ, Chen LX, Kang N, Hayashi T, Tashiro S, Onodera S, Qiu F, Ikejima T (2014) Nitric oxide induces apoptosis and autophagy; autophagy down-regulates NO synthesis in physalin A-treated A375-S2 human melanoma cells. Food Chem Toxicol 71:128–135
Jiang YY, Yang R, Wang HJ, Huang H, Wu D, Tashiro S, Onodera S, Ikejima T (2011) Mechanism of autophagy induction and role of autophagy in antagonizing mitomycin C-induced cell apoptosis in silibinin treated human melanoma A375-S2 cells. Eur J Pharmacol 659:7–14
Kang YJ, Chung HJ, Nam JW, Park HJ, Seo EK, Kim YS, Lee D, Lee SK (2011) Cytotoxic and antineoplastic activity of timosaponin A-III for human colon cancer cells. J Nat Prod 74:701–706
Kanter-Lewensohn L, Dricu A, Girnita L, Wejde J, Larsson O (2000) Expression of insulin-like growth factor-1 receptor (IGF-1R) and p27Kip1 in melanocytic tumors: a potential regulatory role of IGF-1 pathway in distribution of p27Kip1 between different cyclins. Growth Factors 17:193–202
Kimmelman AC (2011) The dynamic nature of autophagy in cancer. Genes Dev 25:1999–2010
Kundu M, Thompson CB (2008) Autophagy: basic principles and relevance to disease. Annu Rev Pathol Mech 3:427–455
Liu Y, Yang Y, Ye Y-C, Shi Q-F, Chai K, S-i Tashiro, Onodera S, Ikejima T (2012) Activation of ERK-p53 and ERK-mediated phosphorylation of bcl-2 are involved in autophagic cell death induced by the c-Met inhibitor SU11274 in human lung cancer A549 cells. J Pharmacol Sci 118:423–432
Nilsson C, Aboud S, Karlen K, Hejdeman B, Urassa W, Biberfeld G (2008) Optimal blood mononuclear cell isolation procedures for gamma interferon enzyme-linked immunospot testing of healthy Swedish and Tanzanian subjects. Clin Vaccine Immunol 15:585–589
Qin J, Xin H, Nickoloff BJ (2012) Specifically targeting ERK1 or ERK2 kills melanoma cells. J Transl Med 10:15
Siegfried Z, Bonomi S, Ghigna C, Karni R (2013) Regulation of the Ras-MAPK and PI3K-mTOR signalling pathways by alternative splicing in cancer. Int J Cell Biol 2013:568931
Suzuki K, Hino M, Kutsuna H, Hato F, Sakamoto C, Takahashi T, Tatsumi N, Kitagawa S (2001) Selective activation of p38 mitogen-activated protein kinase cascade in human neutrophils stimulated by IL-1beta. J Immunol 167:5940–5947
Sy LK, Yan SC, Lok CN, Man RY, Che CM (2008) Timosaponin A-III induces autophagy preceding mitochondria-mediated apoptosis in HeLa cancer cells. Cancer Res 68:10229–10237
Tsai CH, Yang CW, Wang JY, Tsai YF, Tseng LM, King KL, Chen WS, Chiu JH, Shyr YM (2013) Timosaponin AIII suppresses hepatocyte growth factor-induced invasive activity through sustained ERK activation in breast cancer MDA-MB-231 cells. Evid Based Complement Alternat Med 2013:421051
Wang AX, Qi XY (2013) Targeting RAS/RAF/MEK/ERK signaling in metastatic melanoma. IUBMB Life 65:748–758
Yang J, Wu LJ, Tashino S, Onodera S, Ikejima T (2008a) Reactive oxygen species and nitric oxide regulate mitochondria-dependent apoptosis and autophagy in evodiamine-treated human cervix carcinoma HeLa cells. Free Radic Res 42:492–504
Yang J, Wu LJ, Tashiro S, Onodera S, Ikejima T (2008b) Nitric oxide activated by p38 and NF-kappaB facilitates apoptosis and cell cycle arrest under oxidative stress in evodiamine-treated human melanoma A375-S2 cells. Free Radic Res 42:1–11
Yu Y, Fan SM, Yuan SJ, Tashiro S, Onodera S, Ikejima T (2012) Nitric oxide (*NO) generation but not ROS plays a major role in silibinin-induced autophagic and apoptotic death in human epidermoid carcinoma A431 cells. Free Radic Res 46:1346–1360
Zhang J, Peng B, Chen X (2005) Expressions of nuclear factor kappaB, inducible nitric oxide synthase, and vascular endothelial growth factor in adenoid cystic carcinoma of salivary glands: correlations with the angiogenesis and clinical outcome. Clin Cancer Res 11:7334–7343
Acknowledgments
Financial supported by National Natural Science Foundation of China (81373925) and Program for Liaoning Excellent Talents in University (LR2012034) are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, Y., Xu, L., Lou, LL. et al. Timosaponin AIII induces apoptosis and autophagy in human melanoma A375-S2 cells. Arch. Pharm. Res. 40, 69–78 (2017). https://doi.org/10.1007/s12272-016-0763-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0763-3