
Abstract
Two new ursane-type triterpenoid saponins, bodiniosides M (1) and N (2), along with three known saponins, oblonganosides I (3), pseudobuxussaponin B (4) and bodinioside A (5), were isolated from the aerial parts of Elsholtzia bodinieri. The structures of compounds 1 and 2 were characterized by spectroscopic data as well as acid hydrolysis and GC analysis as 3-O-β-d-xylopyranosyl-19α-hydroxy-23-acetoxy-urs-12(13)-en-28-oic acid 28-O-α-l-rhamnopyranosyl-(1-2)-β-d-glucopyranoside and 3-O-β-d-glucopyranosyl-2α,19α-dihydroxy-urs-12(13)-en-28,20β-lactone. Compounds 1 and 5 exhibited potent anti-HCV activities in vitro with a selective index of 6.53 and 4.41, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A species of the genus Elsholtzia (Labiatae), E. bodinieri Van’t is an annual herbaceous plant widely found in the mountainous area of the west and southwest districts of China (Chinese name “Dongzisu”), that has been used as an herbal tea or traditional folk medicine for the prophylaxis and treatment of cough, headache, pharyngitis, fever, and hepatitis (Jiangsu New Medical College 1985). Previous studies of E. bodinieri root have led to the isolation of triterpenoid saponins (Hu et al. 2007a; Zhu et al. 2002; Li et al. 2005), sesquiterpene glycosides (Hu et al. 2007b), clerodane diterpenoid glycosides (Hu et al. 2008), and phenolic compounds (Hu et al. 2007c). As a part of an investigation on the chemical components of herbal tea, we previously reported the isolation of flavonoid glycosides (Li et al. 2008) and triterpenoid saponins (Li et al. 2005, 2012b; Zhao et al. 2015) from the aerial parts of this plant. As a continuation of this work, we further systematically investigated the chemical components of the aerial parts of the plant, leading to the isolation of two new ursane-type triterpenoid saponins, bodiniosides M (1) and N (2), along with three known saponins, oblonganoside I (3) (Wu et al. 2007), pseudobuxussaponin B (4) (Taketa and Schenkel 1994), and bodinioside A (5) (Li et al. 2005). In the present paper, we mainly report the isolation and structural elucidation of compounds 1 and 2 (Fig. 1).

The structures of compounds 1 and 2
Materials and methods
General experimental procedures
Optical rotations were obtained using a Jasco DIP-370 digital polarimeter. UV spectra were run on a UV-210A spectrophotometer. IR spectra were recorded on a Bio-Rad FtS-135 spectrophotometer in a KBr disk. 1D and 2D NMR spectra were recorded using Bruker AM-400 and DRX-500 instruments with tetramethylsilane (TMS) as an internal standard. EIMS were measured on a VG Auto-Spec-3000 spectrometer. ESIMS and HRESIMS were obtained with an API-Qstar-TOF instrument. GC analysis was run on Agilent Technologies HP5890 gas chromatograph with flame ionization detector. Semi-preparative HPLC was performed on an Agilent 1200 liquid chromatograph with a ZORBAX SB-C18 (5 mm, 9.4 mm × 250 mm) column. Column chromatography (CC) was carried out on silica gel (200–300 mesh, 100–200 mesh, 80–100 mesh, Qingdao Marine Chemical Factory, Qingdao, China), Lichroprep RP-18 (43–63 µm, Merck, Darmstadt, Germany) and Sephadex LH-20 (Amersham Biosciences AB, Uppsala, Sweden). Fractions were monitored by TLC plates (Si gel G, Qingdao Marine Chemical Factory, Qingdao, China), and spots were visualized by heating silica gel plates sprayed with 5 % H2SO4-EtOH.
Materials
The aerial parts of E. bodinieri were collected from Honghe, Yunnan Province, P. R. China, in May 2010, and identified by Prof. Hai-Zhou Li, Kunming University of Science and Technology. A voucher specimen (KMUST 20100501) was deposited at the Laboratory of Phytochemistry, Faculty of Life Science and Technology, Kunming University of Science and Technology. Ribavirin (99 %) and gentamycin (607 IU/mg, anhydrous) were purchased from Shandong Phoenix Pharmaceutical Co., Ltd., and Apollo Scientific Ltd, respectively. The purity (>95 %) of compounds 1, 2, 3 and 5 used for biological assay was determined by HPLC.
Extraction and isolation
The dried aerial parts of E. bodinieri (15 kg) were powdered and extracted with 75 % aq. Me2CO (3 × 35 L, 24 h, each) at room temperature, then concentrated in vacuo to yield an extract, which was suspended in H2O, and successively partitioned with CHCl3, EtOAc, and n-BuOH. The EtOAc extract (507.0 g) was chromatographed over silica gel CC eluting with CHCl3/Me2CO (gradient 1:0~0:1, each 4 L) to afford six fractions, A–F. Fr. F (CHCl3/Me2CO 0:1, 65.5 g) was separated by Sephadex LH-20 gel column (eluted with 30, 60, and 90 % MeOH/H2O) to obtain subfractions F-1~F-4. Fr. F-1 (12.5 g) was isolated by RP-18 CC (eluted with 30, 60, and 90 % MeOH/H2O) to get subfractions F-1-1~F-1-6. Fr. F-1-6 (1.4 g) was subjected to silica gel CC (eluted with CHCl3/MeOH/H2O 8: 2: 0.5–6.5: 3.5: 1) to yield F-1-6-1~F-1-6-5. Compounds 2 (15.9 mg) and 3 (21.5 mg) were obtained from Fr. F-1-6-2 (157.0 mg) by silica gel CC eluted with CHCl3/MeOH/H2O 8: 2: 0.5–6.5: 3.5: 1. Fr. F-1-6-3 (225.0 mg) was purified over semi-preparative HPLC by using (65:35) as the mobile phase (3 mL/min) MeOH/H2O to give compounds 4 (tR = 21.5 min, 15.9 mg), 5 (tR = 25.0 min, 25.5 mg), and 1 (tR = 28.0 min, 33.6 mg).
Bodinioside M (1): white amorphous powder. \([\alpha ]_{D}^{18.8}\) = −19.89 (c = 0.18, MeOH). IR (KBr): 3441, 2931, 1721, 1632, 1386, 1255, 1074 cm−1; UV λmax (MeOH) nm (logε): 203 (4.8), 258 (2.8); ESI-MS (neg.) m/z: 1005 [M + Cl]−); HR-ESI-MS (neg.) m/z: 969.5055 [M-H]− (Calcd for C49H77O19, 969.5059). 1H and 13C-NMR, see Table 1.
Bodinioside N (2): white amorphous powder. \([\alpha ]_{D}^{18.2}\) = −29.25 (c = 0.08, MeOH). IR (KBr): 3441, 2931, 1719, 1638, 1389, 1248, 1067 cm−1; UV λmax (MeOH) nm (log ε): 202 (3.4); ESI-MS (pos.) m/z: 687 [M + Na]+, 703 [M + K]+; HR-ESI-MS (pos.) m/z: 687.3701 [M + Na–H]+ (Calcd for C36H56O11Na, 687.3720). 1H and 13C-NMR, see Table 1.
Acid hydrolysis for sugar analysis
A solution of 1 and 2 (1.0 mg for each compound) in 1 M HCl (0.4 mL) was heated at 90–100 °C in a screw-capped vial for 5 h. The mixture was neutralized by addition of Amberlite IRA400 (OH− form) and filtered. The filtrate was dried in vacuo, dissolved in 0.2 mL of pyridine containing l-cysteine methyl ester (10 mg/mL) and reacted at 60 °C for 1 h. A solution (0.2 mL) of trimethylsilylimidazole in pyridine (10 mg/mL) was added to this mixture, and it was heated at 60 °C for 1 h. The final mixture was directly analyzed by GC [30QC2/AC-5 quartz capillary column (30 m × 0.32 mm) with the following conditions: column temperature: 180 °C/280 °C; programmed increase 3 °C/min; carrier gas: N2 (1 mL/min); injection and detector temperature: 250 °C; injection volume: 4 µL; split ratio: 1/50]. The standards were prepared following the same procedure. Under these conditions, the retention times of d- and l-glucose, d- and l-xylose, and l-rhamnose were 18.29, 18.87, 13.35, 14.01, and 14.97 min, respectively. During co-injection studies, identical retention times were observed between the different hydrolysates and authentic standards.
Anti-HCV activity
Inhibitor preparation. For the inhibitory activity assays, compounds 1, 2, 3, and 5 were dissolved and then serially diluted with DMSO, using DMSO and ribavirin as blank and positive controls, respectively.
Cell line and cell culture. Stocks of infectious hepatitis C virus (HCV) J6/JHH-1 viral particles were generated as previously described (Li et al. 2012a) and aliquoted for storage at −80 °C. The HCV load (RNA copies) in stocks was measured with real-time quantitative RT-PCR.
Anti-HCV assay in vitro. Anti-HCV assay was carried out in clear-bottomed 96-well plates. Huh7.5.1 cells were seeded at a density of 1.5 × 104 cells/well in 100 µL of DMEM culture medium and incubated overnight, and virus stocks were added into the wells and incubated for 8 h. Before the samples were added, the supernatants were removed and cells were washed with completed medium 5 times. The samples or controls were serially diluted in DMEM and then added to the appropriate wells. After incubation for 3 days, the supernatants were collected to determine their viral load with real-time quantitative RT-PCR.
Cytotoxicity assay. The toxicities of the compounds were assayed by a modified MTT method (Li et al. 2012a). In brief, the test samples were prepared at different concentrations. After Huh7.5.1 cells had been seeded in a 96-well microplate for 4 h, the samples (20 μL) were placed in each well and incubated for 3 days at 37 °C; then, 0.1 mL MTT was added for 4 h. After removal of the MTT medium, DMSO (100 μL/well) was added onto the microplate for 10 min. The formazan crystals were dissolved, and the absorbance was measured on a microplate reader at 490 nm.
Results and discussion
Compound 1 was obtained as a white amorphous powder. Its molecular formula was determined as C49H78O19 according to the [M–H]− peak at m/z 969.5055 in the negative HR-ESI-MS, indicating eleven degrees of unsaturation. It exhibited UV maximum absorption at 203 and 258 nm. Its IR spectrum showed absorption typical of OH (3441 cm−1) and carbonyl group (1721 cm−1).
The 1H NMR spectrum (Table 1) showed signals of five singlet methyls at δ H 0.77, 0.81, 1.00, 1.19, and 1.32 (each 3H, s), one doublet methyl signal at δ H 0.92 (3H, d, J = 6.7 Hz), one proton in oxygen-bearing carbon at δ H 3.56 (1H, m, H-3), one oxygenated CH2 proton at δ H 3.81 (1H, m, H-23a), 3.15 (1H, m, H-23b), and one olefinic proton at δ H 5.30 (1H, br. s, H-12) in the aglycone moiety. Except the signals for the aglycone, the 1H NMR spectrum of compound 1 also showed three signals for anomeric protons at δ H 5.43 (1H, s), 5.36 (1H, d, J = 7.6 Hz) and 4.20 (1H, d, J = 7.6 Hz), suggesting the presence of three sugar moieties. The 13C NMR and DEPT spectra of 1 exhibited a total of 49 signals. Among them, 30 signals were attributed to be the aglycone moieties as follows: six methyls, nine methylenes, six methines, and nine quaternary carbons, including one CO groups at δ C 178.4 (C-28), one oxygenated quaternary carbon at δ C 73.6 (C-19), one oxygenated CH group at δ C 83.6 (C-3), and two olefinic carbons at δ C 129.6 (C-12) and 139.5 (C-13). And the remaining 19 signals were assigned as three sugar moieties and an acetoxy group due to signals of δ C 172.7, 20.9 and δ H 2.06 (3H, s). A methine proton signal at δ H 2.48 (δ H, s, H-18) belonging to the aglycone moiety was correlated to carbon signals at δ C 129.6 (C-12), 139.5 (C-13), 43.1 (C-14), 26.5 (C-16), 48.9 (C-17), 73.6 (C-19), and 178.4 (C-28) in the HMBC spectrum, suggesting that this proton could be assigned to H-18 of 19-oxygenated ursane-type triterpene. The above evidences supported that the aglycone of 1 was 3,19-dihydroxyurs-12-en-28-oic acid (Wu et al. 2007; Song et al. 2013).
Acid hydrolysis of 1 with 1 M HCl produced d-glucose (Glc), D-xylose (Xyl) and l-rhamnose (Rha) as sugar residues, which were confirmed by GC analysis of their corresponding trimethylsilylated l-cysteine derivatives. Since NMR signals of three sugar units have undesirable overlapped effects, the HMQC-TOCSY experiment was successfully used to distinguish and assign the 1H and 13C NMR signals of each sugar moiety. The correlations from the anomeric proton signal at δ H 4.20 to five carbon signals at δ C 107.0 (anomeric carbon), 75.3, 78.0, 71.9, and 66.4, as well as from five proton signals at δ H 4.20, 3.16, 3.25, 3.92, and 4.10 to the anomeric carbon, suggested the presence of d-xylopyranose. In a similar way, the 1H and 13C NMR signals for d-glucopyranosyl and l-rhamnopyranosyl were assigned (Table 1). The β anomeric configurations for the xylopyranosyl and glucopyranosyl were determined by means of their 3 J H1,H2 coupling constants (3 J = 7.6 and 7.6 Hz, respectively). Comparison of the 13C chemical shifts deduced the α anomeric configuration for l-rhamnopyranosyl (Li et al. 2012b).
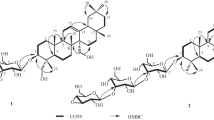
The sequence of the glycoside chains connected to C-3 and C-28 was established by analysis of the HMBC correlations. The absence of any glycosidation shift for Xyl suggested that Xyl was the singlet sugar unit attached at C-3 of the aglycone, which were further confirmed by HMBC correlation of HXyl-1 (δ H 4.20) of C-3 (Fig. 2). A series of HMBC correlations from HGlc-1 (δ H 5.36) to C-28, and from HRha-1 (d, 5.43) to CGlc-2 (δ c 76.5), unambiguously clarified the sequence of the second bidesmosidic part at C-28. In addition, HMBC correlation between H-23 (δ H 3.81, 3.15) and the carbonyl carbon (δ C 172.7) suggested that the acetoxy group was attached at C-23.

Key HMBC correlations of compounds 1 and 2
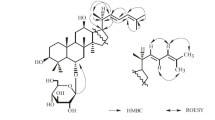
The relative configuration of 1 was derived from ROESY spectrum. As for ursane-type triterpenoid, Me(24), Me(25), Me(26), and C(29) were biogenically assumed to be β-orientations, and H-C(5), H-C(9), Me(23), Me(27) and Me(30) were α-orientations. The NOE correlations between H-3, and Hb-23 with H-C(5) (δ(H) 1.12) in the ROESY spectrum indicated the α-configuration for H-C(3) and Hb-C(23). The α-orientation of the 19-OH was deduced from the β-orientation of Me-29.
Based on the above evidence, the structure of compound 1 was established as 3-O-β-d-xylopyranosyl-19α-hydroxy-23-acetoxy-urs-12(13)-en-28-oic acid 28-O-α-l- rhamnopyranosyl-(1-2)-β-d-glucopyranoside, a new ursane-type saponin, and named as bodinioside M.
Compound 2 was obtained as white amorphous powder. Its molecular formula was deduced to be C36H56O11 by the HR-ESI-MS at m/z 687.3701 [M + Na]+ (Calcd for C36H56O11Na, 687.3720), indicating nine degrees of unsaturation. The IR spectrum exhibited absorptions at 3436 and 1735 cm−1, indicating the existence of hydroxyl and δ-lactone. Its UV spectrum exhibited maximum absorption at 202 nm. The 1H NMR spectrum of 2 showed signals attributable to one oxygenated methine [δ H 3.29 (d, J = 9.25), H-3], one oxygenated methylene [δ H 4.33, m, H-30], one olefinic proton [δ H 6.16 (br. s), H-12], and six tertiary methyl groups (δ H 0.77, 0.92, 1.07, 1.22, 1.40, and 1.60). The 13C NMR and DEPT spectra revealed 36 signals including a lactone carbonyl carbon (δ C 178.8), a pair of olefinic carbons (δ C 135.7 and 125.9), seven quaternary carbons (two oxygenated ones, δ C 87.4 and 73.3), ten methines (seven oxygenated ones, δ C 66.8, 95.6, 106.6, 75.7, 78.8, 71.7, and 78.6), ten methylenes (three oxygenated ones, δ C 63.4, 62.7), and six methyl carbons (δ C 28.5, 18.1, 16.9, 16.2, 23.5, and 26.0). A pile of signals for oxygenated methylenes in the NMR spectra, together with the HSQC correlation of δ H 4.98 (1H, d, J = 7.8) with δ C 106.6 (d), suggested that 2 contained one sugar residue. These NMR spectral data ascribed to the aglycone moiety closely resembled those of glutinsalactone B, an ursane-type triterpene with a δ-lactone ring between C-20 and C-28 (Zhang et al. 2013; Ouyang et al. 1996; Errington and Jefferies 1988).
In comparison of the NMR data of 2 to those of glutinsalactone B, the main difference was the presence of the sugar residue and an additional oxygenated methylene in 2 and the absence of a methylene in glutinsalactone B. The above difference suggested that 2 was glycoside of glutinsalactone B. Acid hydrolysis of 2 with 1 M HCl produced d-glucose as sugar residue as determined by GC analysis. Meanwhile, the anomeric configuration of d-glucose residue was deduced to be β based on the coupling constant of anomeric proton (J = 7.8). The β-d-glucose residue was deduced to be attached at C-3 due to the HMBC correlation of anomeric proton at δ H 4.98 (1H, d, J = 7.8) with C-3. A hydroxyl was assigned to connect to the C-2 on the basis of HMBC correlations from H-1 and H-3 to C-2, thus leading to the presence of the additional oxygenated methylene in 2. A series of HMBC correlations, in which the proton signals at δ H 1.07 (s, H-24) correlated with δ C 95.6 (C-3), δ C 55.8 (C-5), and δ C 40.8 (C-4), δ H 4.33 (H-30) correlated with δ C 87.4 (C-20), δ C 73.3 (C-19), and δ C 25.4 (C-21), and δ H 1.60 (s, H-29) correlated with δ C 50.6 (C-18), 73.3 (C-19), and 87.4 (C-20) further confirmed the correctness of the inference.
The relative configuration of 2 was determined based on a ROESY experiment and comparison of chemical shifts with of those of analogues. ROESY correlations of H-C(2)/Me(24), H-C(2)/Me(25), H-C(18)/H-C(29) indicated the α-hydroxy group at C-2 and C-19. While, the ROESY correlations of H-C(3)/H-C(5) and H-C(3)/Me(23) suggested a β-hydroxy group at C-3. The configuration of the 28, 20β-lactone was determined by comparison with the known triterpene compound (Errington and Jefferies 1988; Ouyang et al. 1996). Thus, the structure of compound 2 was elucidated to be 3-O-β-d-glucopyranosyl-2α,19α-dihydroxy-urs-12(13)-en-28,20β-lactone, and named bodinioside N.
The anti-HCV activity of compounds 1, 2, 3, and 5 were evaluated, and their cytotoxicity was measured in parallel with the determination of antiviral activity, using ribavirin as positive control (CC50 = 95.94 nM, EC50 = 9.57 nM). Among the compounds tested, compound 1 exhibited potent anti-HCV activity, with an EC50 value of 11.50 nM, a CC50 value of 75.15 nM, and a SI value of 6.53. Moreover, compound 5 showed moderate anti-HCV activity with an SI value of 4.41 (Table 2). This study suggests that the triterpenoid saponins isolated from E. bodinieri exhibit potent anti-HCV activities in vitro.
The isolated compounds (1–5) were evaluated for anti-HIV-1 effects and anti-influenza A virus activities, but all were found to be inactive.
References
Errington SG, Jefferies PR (1988) Triterpenoid sapogenins of Pittosporum hillyraeoides. Phytochemistry 27(2):543–545
Hu HB, Zheng XD, Hu HS, Jian YF (2007a) Triterpenoid saponins from Elsholtzia bodinieri. Bull Korean Chem Soc 28(9):1519–1522
Hu HB, Jian YF, Zheng XD, Cao H (2007b) Three sesquiterpene glycosides from Elsholtzia bodinieri. Bull Korean Chem Soc 28(3):467–470
Hu HB, Ean YF, Cao H, Zheng XD (2007c) Phenolic compounds from Elsholtzia bodinieri Van’t. J Chin Chem Soc 54(5):1189–1194
Hu HB, Cao H, Jian YF, Zheng XD, Liu JX (2008) Two new clerodane diterpenoid glucosides and other constituents from the roots of Elsholtzia bodinieri. Indian J Chem B: Org 47:166–170
Jiangsu New Medical College (1985) The Dictionary of Chinese Medicine. Shanghai Press of Science and Technology
Li RT, Li JT, Wang JK, Han QB, Zhu ZY, Sun HD (2005) Two new E-secoursane glycosides: bodiniosides A and B, isolated from Elsholtzia bodinieri. Helv Chim Acta 88:252–258
Li RT, Li JT, Wang JK, Han QB, Zhu ZY, Sun HD (2008) Three new flavonoid glycosides isolated from Elsholtzia bodinieri. Chem Pharm Bull 56(4):592–594
Li HM, Tang YL, Zhang ZH, Liu CJ, Li HZ, Li RT, Xia XS (2012a) Compounds from Arnebia euchroma and their related anti-HCV and antibacterial activities. Planta Med 78:39–45
Li HZ, Fu LZ, Li HM, Li RT, Deng XL (2012b) Two new oleanane triterpenoid saponins from Elsholtzia bodinieri. Phytochem Lett 5:572–575
Ouyang MA, Wang HQ, Chen ZL, Yang CR (1996) Triterpenoid glycosides from Ilex kudincha. Phytochemistry 43(2):443–445
Song YL, Zeng KW, Shi XT, Jiang Y, Tu PF (2013) Sibiricasaponins A-E, five new triterpenoid saponins from the aerial parts of Polygala sibirica L. Fitoterapia 84:295–301
Taketa ATC, Schenkel EP (1994) Saponins from Ilex pseudobuxus. Acta Farm Bonaerense 13:159–164
Wu ZJ, Ouyang MA, Wang CZ, Zhang ZK, Shen JG (2007) Anti-Tobacco mosaic virus (TMV) triterpenoid saponins from the leaves of Ilex oblonga. J Agric Food Chem 55(5):1712–1717
Zhang YL, Feng WS, Zheng XK, Cao YG, Lv YY, Chen HH, Kuang HX (2013) Three new ursane-type triterpenes from the leaves of Rehmania glutinosa. Fitoterapia 89:15–19
Zhao XW, Zhong JD, Li HM, Li RT (2015) Three new 18,19-seco-ursane glycosides from Elsholtzia bodinieri. Phytochem Lett 12:308–312
Zhu WM, He HP, Wang S, Zuo GY, Hao XJ (2002) Two new triterpenoid glycosides from Elsholtzia bodinieri Van’t. Chin Chem Lett 13(3):253–255
Acknowledgments
We gratefully acknowledge the financial support for this research work by the National Natural Science Foundation of China (No. 21262021, 21572082), the State Key Laboratory of Phytochemistry and Plant Resources in West China (No. P2015-KF01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhong, JD., Zhao, XW., Chen, XQ. et al. Two new ursane-type triterpenoid saponins from Elsholtzia bodinieri . Arch. Pharm. Res. 39, 771–777 (2016). https://doi.org/10.1007/s12272-016-0750-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0750-8