
Abstract
A phytochemical investigation for the rhizome extract from Sinomenium acutum (Menispermaceae) resulted in the isolation of several active principles responsible for the anti-osteoclastogenic property of the extract, together with related isoquinoline alkaloids (1–13) including two new compounds, 1 and 2. Among isolated compounds, salutaridine (7), dauricumine (10), cheilanthifoline (12), and dauriporphine (13) were observed to give significant inhibitions on receptor activator of nuclear factor-κB ligand-induced differentiation of mouse bone marrow-derived macrophages into multinucleated osteoclasts, respectively. The chemical structures of two newly isolated compounds, 1 and 2 were established as 8-demethoxycephatonine (1) and 7(R)-7,8-dihydrosinomenine (2), by spectroscopic analyses including 2D NMR experiments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The species, Sinomenium acutum (Menispermaceae) is a deciduous climbing plant native to Korea, Japan and China. The rhizomes of the species have been used frequently to treat rheumatic and arthritic diseases in traditional Chinese medicine (Zhao et al. 2012). It is also regarded as a valuable source of plant alkaloids with diverse skeletons, such as morphinane, hasubanane and aporphine alkaloids. Especially, sinomenine (4), a representative alkaloidal component of the species has been reported to exhibit a variety of biological activities including anti-arthritic (Zhou et al. 2008), anti-inflammatory (Wang and Li 2011), and immunosuppressive properties (Wang and Li 2011). In addition, it has been paid much attention by chemists and pharmacologists, because it was reported recently to suppress osteoclast formation and bone loss (Li et al. 2013).
In the course of investigating for anti-osteoclastogenic effects of the traditional herbal medicines in Korea, the methanol extract from the rhizomes of S. acutum was found to exhibit a significant inhibition on the receptor activator of nuclear factor-κB ligand (RANKL)-induced differentiation of mouse bone marrow-derived macrophages (BMMs) into tartrate-resistant acid phosphatase-positive multinucleated cells (TRAP+-MNCs; Fig. S1). It was generally accepted that the balance between osteoclastic bone resorption and osteoblastic bone formation preserved skeletal homeostasis in adults (Walsh et al. 2006), but an imbalance caused by over-activated osteoclasts and/or their increased number consequently led to osteoclast-related bone diseases such as osteoporosis and rheumatoid arthritis (Manolagas 2000). Osteoclasts are large MNCs belonging to the monocyte/macrophage lineage of hematopoietic cells (Suda et al. 1999). Two essential cytokines, macrophage colony-stimulating factor-1 (M-CSF) and RANKL triggered the differentiation of osteoclast precursors into TRAP+-MNCs with bone resorbing activity (Boyle et al. 2003).
For the purpose of identifying active principles of the extract from the rhizomes of S. acutum, which were responsible for the inhibitory effect on the RANKL-induced differentiation of BMMs into TRAP+-MNCs, extensive phytochemical studies on the extract were performed, which finally resulted in the isolation of four active components such as salutaridine (7), dauricumine (10), cheilanthifoline (12), and dauriporphine (13).
Materials and methods
General experimental procedures
Optical rotations were recorded on a Rudolph Autopol IV polarimeter. The high resolution electrospray ionization (HREIMS) and electron impact mass spectra were obtained using a Q-Tof micro LC–MS/MS instrument (Waters, USA) and CP3800-1200L (Varian, USA) mass spectrometer, respectively. NMR spectra were obtained by a Bruker AM 500 spectrometer using TMS as an internal standard for 1H NMR, 13C NMR, DEPT, COSY, HMQC, HSQC, HMBC and NOESY. Preparative-HPLC was performed on a Futecs P-4000 system with a Shim-pack prep-ODS(H) kit column (5 µm, 20 mm × 25 cm). Isolation and purification was carried out using a medium-pressure liquid chromatographic system [Buchi pump Module C-601, silica gel 60 (230–400 mesh, Merck), ODS (Cosmosil 140 C18)].
Plant material
The rhizomes of S. acutum were purchased at Gyeongdong market (Korea) and identified by Dr. Young sup Kim. A voucher specimen (KR1316) was deposited at the herbarium of the Korea Research Institute of Chemical Technology (KRICT).
Extraction and isolation
The dried rhizomes (6 kg) of S. acutum were soaked twice in 90 l of methanol at room temperature for 7 days. The concentrated methanol extract (737 g) was suspended in 4 L of distilled water and adjusted to pH 12.0 with 1 N NaOH, and then extracted sequentially with equal volume n-butanol which gave 156 g of an alkaloids-rich fraction. The alkaloids-rich fraction (156 g) was dissolved in EtOAc and allowed to crystallize at room temperature to give compound 4 (5.5 g). And then, the remained alkaloids-rich fraction was fractionated into five sub-fractions (Fr.1–5) using silica gel column chromatography and gradient elution with dichloromethane/MeOH (100:1–1:1). Fr.1 was subjected to column chromatography with NH2 gel (Merck Lichroprep NH2) eluted with a n-hexane:EtOAc (50:1–3:1) to give compound 13 (3.1 mg). Fr.2 was further purified by prep-HPLC (30–90 % MeOH) to yield compounds 3 (37.1 mg), 1 (29.1 mg), 12 (6.0 mg). Fr.3 was repeatedly subjected to silica gel column chromatography (dichloromethane:MeOH = 50:1–1:1) and purified using prep-HPLC with 30–90 % MeOH gradient to afford compounds 5 (23.8 mg), 7 (37.2 mg), 8 (58.7 mg), 9 (7.4 mg), and 10 (9.3 mg). Fr.4 was subjected to column chromatography with NH2 gel (Merck Lichroprep NH2) eluted with a n-hexane:EtOAc (50:1–3:1) to give compounds 6 (92.4 mg) and 2 (15.2 mg). Fr.4 was further purified through Sephadex LH-20 chromatography and prep-HPLC (10–70 % MeOH) to yield compound 11 (2.1 g).
8-Demethoxycephatonine (1)
Yellow amorphous powder; \([\alpha ]_{D}^{20}\) −141.5 (c 1.00, CH3OH); 1H NMR (CDCl3, 500 MHz); 13C NMR (CDCl3, 125 MHz) (Table 1); HREIMS m/z 329.1626 [M+Na]+ (calcd for C19H23NO4, 329.1627).
7(R)-dihydrosinomenine (2)
Yellow amorphous powder; \([\alpha ]_{D}^{20}\) +37.6 (c 0.33, CH3OH); 1H NMR (CDCl3, 500 MHz); 13C NMR (CDCl3, 125 MHz) (Table 2); HREIMS m/z 331.1776 [M+Na]+ (calcd for C19H25NO4, 331.1784).
Hydrogenation of 4
Compound 4 (0.1 g) was dissolved in MeOH with 10 % Pd/C (0.01 g) and then stirred for about 72 h at the room temperature. After filtration, the reaction mixture was concentrated to dryness and purified by column chromatography on silica gel eluted with dichloromethane:MeOH (5–95 % MeOH gradient) to yield 62.1 mg of 7(R)-dihydrosinomenine and 24.5 mg of 7(S)-dihydrosinomenine.
Cell cultures and osteoclast differentiation
This study was carried out in strict accordance with the recommendations in the Standard Protocol for Animal Study of KRICT (No. 2012-7D-02-01). The protocol (ID No. 7D-M1) was approved by the Institutional Animal Care and Use Committee of KRICT. All efforts were made to minimize suffering. In detail, bone marrow-derived cells (BMCs) were obtained from femur and tibia of 5–6-week-old male ICR mice (Damool Science, Daejeon, Korea) by flushing femurs and tibias with α-MEM supplemented with antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin; Invitrogen, NY, USA). BMCs were cultured in a culture dish in α-MEM supplemented with 10 % fetal bovine serum (Invitrogen, NY, USA) with 10 ng/ml of mouse recombinant M-CSF (R&D Systems, MN, USA) for 1 day. After 1 day, non-adherent BMCs were plated on a Petri dish and cultured for 3 days in the presence of M-CSF (30 ng/ml). After non-adherent cells were washed out, adherent cells were used as BMMs. For osteoclastogenesis, BMMs were cultured in the presence of 10 ng/ml of mouse recombinant RANKL (R&D Systems) and M-CSF (30 ng/ml) for 4 days to differentiate them into mature TRAP-positive multinucleated osteoclast cells (TRAP+-MNCs). BMMs were pre-treated with compound for 1 h before RANKL treatment.
TRAP staining and activity assay
Cells were fixed with 3.7 % formalin for 5 min, permeabilized with 0.1 % Triton X-100 for 10 min, and stained with TRAP solution (Sigma-Aldrich, MO, USA) for 10 min. TRAP+-MNCs (cells with 10 nuclei or more; 10 ≤ N) were counted as mature osteoclasts. For measuring TRAP activity, cells were fixed with 3.7 % formalin for 5 min, permeabilized with 0.1 % Triton X-100 for 10 min, and treated with TRAP buffer (100 mM sodium citrate pH 5.0, 50 mM sodium tartrate) containing 3 mM p-nitrophenyl phosphate (Sigma-Aldrich, MO, USA) at 37 °C for 5 min. Reaction mixtures were transferred into new plates containing an equal volume of 0.1 N NaOH, and optical density values were measured at 405 nm.
Cytotoxicity assay
BMMs were cultured at a density of 1 × 104 cells/well on a 96-well plate in triplicate with M-CSF (30 ng/ml) and tested materials. After 3 days, the cell survival was evaluated by CCK-8 kit according to the manufacturer’s protocol (Dojindo Molecular Technologies, Japan).
Western blot analysis
BMMs (1 × 106 cells/well in a six-well plate) were pre-treated with each four isoquinoline alkaloid (10 μM) 1 h before incubating with RANKL (10 ng/ml) for 5 min. Cells were then washed with ice-cold phosphate buffered saline and lysed in lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 1 % Triton X-100, 1 mM sodium fluoride, 1 mM sodium vanadate, and 1 % deoxycholate) containing protease inhibitors. Lysates were boiled in sodium dodecyl sulfate (SDS) sample buffer for 5 min, subjected to 10 or 12 % SDS-polyacrylamide gel electrophoresis, and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). Transferred PVDF membrane was washed with TBST (10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1 % Tween 20) and incubated with TBST with 5 % skim milk. The membrane was then probed with the primary antibody, washed three times with TBST for 30 min, incubated with secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology, CA, USA). for 2 h, and washed three times for 30 min. Membranes were then developed with SuperSignal west femto maximum sensitivity substrate (Pierce) using the LAS-3000 luminescent image analyzer (Fuji Photo Film Co., Ltd., Japan). Antibody against actin was purchased from Santa Cruz Biotechnology. Antibodies against p-p38, p38, p-JNK, JNK, p-ERK, ERK were purchased from Cell Signaling Technology (MA, USA).
Results
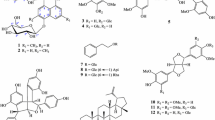
Extensive phytochemical studies were conducted on the rhizomes extract of S. acutum to investigate active ingredients responsible for the anti-osteoclastogenic property, which finally led to the isolation of 13 kinds of isoquinoline alkaloids (1–13) including two new compounds, 1 and 2 (Fig. 1). Among them, 7, 10, 12, and 13 exhibited extremely potent anti-osteoclastogenic activity. In this paper, we briefly describe the inhibitory effects of active components (7, 10, 12, and 13) on the differentiation of BMMs cells into TRAP+-MNCs as well as the structural elucidation of two new isoquinoline alkaloids, 1 and 2 by spectroscopic evidences.

Isolated compounds (1–13) from the rhizome extract of S. acutum
All isolated alkaloids (1–13) were evaluated for the inhibitory effect on osteoclast differentiation. As shown in Fig. 2, four alkaloids (7, 10, 12, and 13) significantly inhibited the RANKL-induced formation of TRAP+-MNCs in a dose-dependent manner, whereas other isolated alkaloids gave poor inhibition below 10 μM concentration. The concentration of 7, 10, 12, and 13 required for the 50 % inhibitory effect (IC50) on osteoclast differentiation was calculated as 3.8, 4.2, 3.5 and 2.8 μM, respectively, as the IC50 value of reference drug, saurolactam was observed as 29 μM (Kim et al. 2009). Besides, none of them (7, 10, 12, and 13) showed any significant cytotoxic effect on BMMs cells below 10 μM, suggesting that the anti-osteoclastogenic activities of them were not described to the potential to inhibit the survival of BMMs. As mentioned previously, sinomenine (4), a major alkaloid of this species has been reported recently to give anti-osteoclastogenic activity and the IC50 value was esteemed about 100 μM (Li et al. 2013). These results were well accorded with our experimental results, i.e., 4 was observed to give excellent activity at 100 μM concentration, but it exhibited negligibly poor activity below 50 μM. On the other hand, compounds 7, 10, 12, and 13, structurally similar to 4 exhibited much potent inhibition upon the RANKL-induced formation of TRAP+-MNCs4 even below 5 μM concentration (Fig. 2). Thus, it is supposed that the overall anti-osteoclastogenic activity of the rhizome extract of S. acutum was predominantly due to the potent activities of 7, 10, 12, and 13, rather than due to the main alkaloid, sinomenine (4).

Effect of isoquinoline alkaloids (7, 10, 12 and 13) on osteoclast differentiation. a BMMs were cultured with RANKL (10 ng/ml) and M-CSF (30 ng/ml) in the presence of the indicated concentrations of isoquinoline alkaloids for 4 days. Cells were fixed in 3.7 % formalin for 5 min, permeabilized in 0.1 % Triton X-100 for 10 min, and stained for TRAP, a marker enzyme of osteoclasts. b TRAP-positive multinuclear cells (nuclei ≥10) were counted as osteoclasts. *P < 0.05, **P < 0.01, ***P < 0.001
As one of possible mechanisms underlying the anti-osteoclastogenic actions of four isoquinoline alkaloids (7, 10, 12, and 13), we investigated whether they can affect the MAP kinase signaling molecules including p38, JNK, and ERK, which are known to play an important role in the early signaling event induced by RANKL. BMMs were pre-treated with each isoquinoline alkaloid (7, 10, 12, and 13) before RANKL treatment, and the cell lysates were analyzed by western blotting. RANKL strongly induced the phosphorylation of all signaling molecules such as p38, JNK, and ERK, but the RANKL-induced phosphorylation of JNK was attenuated by each isoquinoline alkaloid (7, 10, 12, and 13). In addition, the RANKL-induced phosphorylation of ERK and p38 were likely to be attenuated by 12 and 13. These results suggested that the anti-osteoclastogenic activities of 7, 10, 12, and 13 could be in part due to the potential to inhibit the RANKL-induced phosphorylation of all MAP kinases (Fig. 3).

Inhibitory effects of four isoquinoline alkaloids (7, 10, 12 and 13) on RANKL-induced activation of MAP kinases. The effects of four isoquinoline alkaloids on RANKL-induced phosphorylation of MAP kinases were evaluated by western blot analysis. Briefly, BMMs were pre-treated with each four isoquinoline alkaloid (10 μM) 1 h before incubating with RANKL (10 ng/ml) for 5 min. Actin was used as an internal control
As mentioned before, in the course of phytochemical survey, two novel isoquinoline compounds (1 and 2) were isolated from the extract. To the best of our knowledge, 1 and 2 were never been isolated from this species and even from nature.
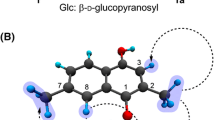
Compound 1 was obtained as a yellow amorphous powder, \([\alpha ]_{D}^{20}\) −141.5 (c 1.00, CH3OH). The molecular formula of 1 was established as C19H23NO4 at m/z 329.1626 (calcd 329.1627) [M+Na]+ by HREIMS. The proton NMR spectrum of 1 was highly similar with that of 3, except that two methoxy signals (δ 3.86, and δ 3.67) were observed in 1, whereas three methoxy signals (δ 3.86, δ 3.85 and δ 3.68) in 3 (Table 1). Besides, the mass difference (Δ −14) between 1 and 3 implied that one of three methoxy groups found in 3 was replaced with a hydroxyl group in 1. A distinct NOE correlation between H-4 and methoxy protons on C-3, and other NOE correlation between H-8 and methoxy protons on C-7 (Fig. 4) suggested that the hydroxyl group was attached at C-2. Thus, the chemical structure of 1 was established as 8-demethoxycephatonine as depicted in Fig. 1.

Key 1H–1H COSY (thick line), HMBC (right pointing arrow) and NOESY (double pointing arrow) correlations of 1 and 2
Compound 2 was obtained as yellow amorphous powder, \([\alpha ]_{D}^{20}\) +37.6 (c 0.33, CH3OH). The molecular formula of 2 was established as C19H23NO4 at m/z 331.1776 (calcd 331.1784) [M+Na]+ by HREIMS, 2 mass units higher than that of 4. The proton NMR spectrum of 2 displayed a high similarity with that of 4. However, a singlet signal observed at δ 5.49 (H-8) of 4 was disappeared, whereas a dd signal at δ 3.93 and additional methylene protons at δ 2.20 and δ 1.64 were observed in 2, which implied that the double bond between C-7 and C-8 of 4 might be reduced to become 2. Thus, all proton and carbon signals of 2 were completely assigned by the aid of 2D NMR experiments such as HSQC, HMBC and NOESY (Table 2). The relative configuration of 2, particularly the chirality of C-7 was established by the NOESY experiment; the signal of H-7 was correlated with H-14, which strongly suggested the methoxy group was attached at C-7 via α-orientation as depicted in Fig. 4. On the basis of these data, the chemical structure of 2 was established as the 7(R)-7,8-dihydrosinomenine as depicted in Fig. 1. On the other hand, the catalytic hydrogenation of sinomenine (4) with Pd/C yielded a pair of stereoisomers, i.e., 7(R)-dihydrosinomenine and 7(S)-dihydrosinomenine as a ratio of 5:2. This is the first report of the isolation of 7(R)-dihydrosinomenine (2) from the nature. Other purified components were identified as 8-demethoxyrunanine (3) (Wang et al. 2007), sinomenine (4) (Kashiwaba et al. 1994), 14-episinomenine (5) (Kashiwaba et al. 1996), sinomenine N-oxide (6) (Bao et al. 2005), salutaridine (7) (Bracher et al. 2004), acutumine (8) (Sugimoto et al. 2001), acutumidine (9) (Sugimoto et al. 2001), dauricumine (10) (Min et al. 2006), magnoflorine (11) (Holzbach and Lopes 2010), cheilanthifoline (12) (Cheng et al. 2008), and dauriporphine (13) (Min et al. 2006) by direct comparison of spectroscopic data with those of published literatures.
In summary, two new isoquinoline alkaloids, 8-demethoxycephatonine (1) and 7(R)-7,8-dihydrosinomenine (2), along with eleven known compounds (3–13) were isolated from the rhizome extract of S. acutum. Among them, salutaridine (7), dauricumine (10), cheilanthifoline (12), and dauriporphine (13) were observed to give significant inhibitions on RANKL-induced differentiation of BMMs into multinucleated osteoclasts, respectively.
Discussion
This is the first report of anti-osteoclastogenic activity of isoquinoline alkaloids isolated from the rhizome extract of S. acutum (7, 10, 12 and 13). Furthermore, the structural information gained from these anti-osteoclastogenic isoquinoline alkaloids may give insights into the design of new therapeutics for the treatment of osteoclast-related disorders. Once structure–activity relationship studies have been conducted, these compounds may lead to promising new drugs for the treatment of osteoporosis, rheumatoid arthritis, and cancer metastasis.
References
Bao GH, Qin GW, Wang R, Tang XC (2005) Morphinane alkaloids with cell protective effects from Sinomenium acutum. J Nat Prod 68:1128–2230
Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423:337–342
Bracher F, Eisenreich WJ, Mühlbacher J, Dreyer M, Bringmann G (2004) Saludimerines A and B, novel-type dimeric alkaloids with stereogenic centers and configurationally semistable biaryl axes. J Org Chem 69:8602–8608
Cheng X, Wang D, Jiang L, Yang D (2008) DNA topoisomerase I inhibitory alkaloids from Corydalis saxicola. Chem Biodivers 5:1335–1344
Holzbach JC, Lopes LM (2010) Aristolactams and alkamides of Aristolochia gigantea. Molecules 15:9462–9472
Kashiwaba N, Morooka S, Kimura M, Murakoshi Y, Toda J, Sano T (1994) Two new morphinane alkaloids from Stephania cepharantha HAYATA (Menispermanceae). Chem Pharm Bull 42:2452–2454
Kashiwaba N, Morooka S, Kimura M, Ono M, Toda J, Suzuki H, Sano T (1996) New morphinane and hasubanane alkaloids from Stephania cepharantha. J Nat Prod 59:476–480
Kim MH, Ryu SY, Choi JS, Min YK, Kim SH (2009) Saurolactam inhibits osteoclast differentiation and stimulates apoptosis of mature osteoclasts. J Cell Physiol 221:618–628
Li X, He L, Hu Y, Duan H, Li X, Tan S, Zou M, Gu C, Zeng X, Yu L, Xu J, Liu S (2013) Sinomenine suppresses osteoclast formation and Mycobacterium tuberculosis H37Ra-induced bone loss by modulating RANKL signaling pathways. PLoS One 8:e74274
Manolagas SC (2000) Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev 21:115–137
Min YD, Choi SU, Lee KR (2006) Aporphine alkaloids and their reversal activity of multidrug resistance (MDR) from the stems and rhizomes of Sinomenium acutum. Arch Pharmacal Res 29:627–632
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ et al (1999) Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev 20:345–357
Sugimoto Y, Babiker HA, Saisho T, Furumoto T, Inanaga S, Kato M (2001) Chlorinated alkaloids in Menispermum dauricum DC: root culture. J Org Chem 66:3299–3302
Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J, Choi Y (2006) Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol 24:33–63
Wang Q, Li XK (2011) Immunosuppressive and anti-inflammatory activities of sinomenine. Int Immunopharmacol 11:373–376
Wang X, Jin H, Li Z, Qin G (2007) 8-Demethoxyrunanine from Sinomenium acutum. Fitoterapia 78:593–595
Zhao XX, Peng C, Zhang H, Qin LP (2012) Sinomenium acutum: a review of chemistry, pharmacology, pharmacokinetics, and clinical use. Pharm Biol 50:1053–1061
Zhou H, Wong YF, Wang J, Cai X, Liu L (2008) Sinomenine ameliorates arthritis via MMPs, TIMPs, and cytokines in rats. Biochem Biophys Res Commun 376:352–357
Acknowledgments
This research was supported by Research Fund of NRF-2012M3A9C and also by a Grant of the Technology Innovation Program (10038744) of Korea Evaluation Institute of Industrial Technology (KEIT), Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors have no conflict of interest to declare.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lee, J.Y., Kim, KJ., Kim, J. et al. Anti-osteoclastogenic effects of isoquinoline alkaloids from the rhizome extract of Sinomenium acutum . Arch. Pharm. Res. 39, 713–720 (2016). https://doi.org/10.1007/s12272-016-0734-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0734-8