
Abstract
Heart failure and myocardial infarction, global health concerns, stem from limited cardiac regeneration post-injury. Myocardial infarction, typically caused by coronary artery blockage, leads to cardiac muscle cell damage, progressing to heart failure. Addressing the adult heart's minimal self-repair capability is crucial, highlighting cardiac regeneration research's importance. Studies reveal a metabolic shift from anaerobic glycolysis to oxidative phosphorylation in neonates as a key factor in impaired cardiac regeneration, with mitochondria being central. The heart's high energy demands rely on a robust mitochondrial network, essential for cellular energy, cardiac health, and regenerative capacity. Mitochondria's influence extends to redox balance regulation, signaling molecule interactions, and apoptosis. Changes in mitochondrial morphology and quantity also impact cardiac cell regeneration. This article reviews mitochondria's multifaceted role in cardiac regeneration, particularly in myocardial infarction and heart failure models. Understanding mitochondrial function in cardiac regeneration aims to enhance myocardial infarction and heart failure treatment methods and insights.
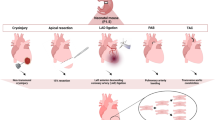
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases (CVD) are the primary cause of mortality from non-communicable diseases worldwide. In 2019, CVD-related deaths reached 17.9 million, accounting for 32% of the total global deaths [1]. Acute myocardial infarction (MI) is precipitated by the rupture of fragile atherosclerotic plaques or concurrent thrombosis, resulting in coronary artery occlusion and progressive necrosis in hypoperfused myocardial regions [2]. Myocardial injury, as in the case of MI, leads to cardiomyocyte loss, fibrotic tissue deposition, and scar tissue formation. This scar tissue, being non-functional, contributes to pathological cardiac remodeling, impairs myocardial contractility, and ultimately leads to heart failure [3,4,5]. As a principal cause of mortality worldwide, effective therapeutic strategies to repair cardiac injury remain elusive. Current clinical interventions primarily aim at early reperfusion of ischemic myocardium through thrombolytic therapy, percutaneous coronary intervention (PCI), and coronary artery bypass graft (CABG) [6]. For patients at high risk following MI, a variety of pharmacological agents are employed as part of guideline-directed medical therapy (GDMT) to reduce mortality and/or the risk of hospitalization due to heart failure. These include β-blockers, angiotensin-converting enzyme inhibitors or angiotensin receptor blockers and mineralocorticoid receptor antagonists [7]. Despite these interventions decelerating the progression of cardiovascular diseases, they do not induce cardiac regeneration. The pursuit of cardiac regeneration therapy, focusing on facilitating myocardial repair and regeneration to restore cardiac function, has emerged as a pivotal area of research. This innovative approach is deemed a potential game-changer in reversing cardiac damage [3].
Traditionally, the heart was considered one of the organs with the least regenerative capacity. However, in 2009, Bergmann et al., through research on C14 isotope that was incorporated into the DNA of human heart muscle cells during the Cold War confirmed that the heart is an organ that is continuously renewing [8]. Although this is a very slow process, insufficient for repairing the heart when it is damaged, this discovery opened the door to the possibility of cardiac regeneration. In recent years, researchers have conducted in-depth exploration of heart regeneration, attempting to find mechanisms that promote myocardial cell proliferation and regeneration. The transition in metabolic patterns has emerged as a pivotal area of interest in this field. While developing cardiac tissue shows substantial regenerative capacity post-injury, mature cardiomyocytes, transitioning from mononuclear diploid to binucleated or polyploid, exit the cell cycle. This metabolic shift experienced by cardiomyocytes postnatally aligns with the halt in cardiac regeneration [9]. Concurrently, in mammals, as the regenerative capability of the heart diminishes, mitochondrial maturation induces a shift from anaerobic glycolysis and lactate production to aerobic glycolysis and oxidative phosphorylation [10]. Among them, the function of mitochondria has gradually gained attention (Table 1).
The high energy demands of the adult heart primarily rely on mitochondrial oxidative phosphorylation, accounting for over 95% of the heart's ATP production [28, 29]. This dynamic organelle also coordinates a range of cellular functions, including signal transduction, calcium regulation, oxidative stress management, and apoptosis [30]. These membrane-bound organelles, originating from endosymbiosis, possess their own genome and a distinctive biochemical reaction network. Their structure comprises the outer membrane (OMM), inner membrane (IMM), cristae, and matrix [31]. The OMM contains vital proteins for mitochondrial and cellular physiology, including translocases that facilitate the transport of mitochondrial precursor proteins and establish physical connections with other cellular regions at membrane contact sites. Additionally, the OMM is instrumental in mitochondrial dynamics [32]. Key pro-inflammatory pathways activated during cell death, including mitochondrial outer membrane permeabilization (MOMP), release damage-associated molecular patterns (DAMPs), partly inciting inflammation [33]. The IMM is intimately involved in energy conversion and apoptosis, acting as a hub for mitochondrial signal transduction [34]. This membrane is the primary site for oxidative phosphorylation, encompassing all complexes pertinent to mitochondrial respiration, such as the mitochondrial respiratory chain and F1-F0 ATP synthase [35]. The mitochondrial cristae, membrane invaginations, are pivotal for oxidative phosphorylation, mitochondrial DNA (mtDNA) , maintenance, and the biogenesis of iron-sulfur clusters [32]. The matrix, housing a small portion of mtDNA encodes essential proteins for mitochondrial respiration [36].
Mitochondria's role in enhancing cardiac regenerative capacity transcends their metabolic functions. They are crucial in maintaining cellular homeostasis and promoting autophagy. Mitochondria are dynamic organelles capable of moving along the cytoskeleton, interacting with other subcellular structures, and shaping their inner and outer membranes through fusion and fission events. Fusion and fission are essential for mitochondrial biogenesis and autophagy [37]. Mitophagy, the selective autophagic degradation of damaged mitochondria, is essential for preserving mitochondrial and cellular homeostasis [38]. In cardiomyocytes, appropriate autophagic activity is critical to maintaining cardiac structural integrity and function [39]. During cardiac diseases like post-myocardial infarction, shifts in mitochondrial dynamics can facilitate cardiomyocyte adaptation and survival, thereby aiding in the repair and regeneration of the afflicted tissue [40].
In summary, there is a close relationship between mitochondrial function and cardiac regeneration, and a deeper comprehension of this relationship provides crucial insights for the treatment of heart failure.
Regulation of ROS
Mitochondria play a crucial role in maintaining cellular energy balance and coping with oxidative stress. Under normal physiological conditions, mitochondria generate a small amount of reactive oxygen species (ROS) [41] through their electron transport chain and oxidative phosphorylation, which participate in cell signaling and maintaining redox balance [42]. However, when ROS production exceeds the cell's antioxidant defense capacity, oxidative stress occurs, causing damage to the cell, such as lipid peroxidation, DNA damage, protein oxidation, irreversible damage to mitochondrial function [43], and reduced ATP generation [44]. Particularly in acute myocardial ischemia, the mechanism of myocardial injury is quite complex, involving the regulation of multiple signaling molecules and pathways. The oxygenation capacity of mitochondria in the ischemic area of the myocardium decreases, inhibiting the respiratory chain, disrupting oxidative phosphorylation, and leading to an increase in ROS levels within the cell. The accumulation of high concentrations of ROS not only disrupts the mitochondrial membrane potential gradient, affecting energy production, but also causes oxidative damage to cardiac myocyte DNA, triggering cell cycle arrest [45, 46].
During the transition from glycolytic metabolism in fetal cardiomyocytes to fatty acid oxidation in adulthood, ROS generated by oxidative phosphorylation may lead to DNA damage and cell cycle arrest, thereby limiting the regenerative capacity of adult myocardium [43]. Studies by Honkoop et al. have found that reprogramming of glycolytic metabolism promotes proliferation and regeneration of injured zebrafish cardiac myocytes [47], and the timing of cell cycle exit and transition in myocardial energy metabolism in neonatal rats coincides with the loss of myocardial regenerative capacity [14, 48]. Therefore, changes in myocardial cell metabolism may trigger entry into the proliferative cycle.
In efforts to promote myocardial regeneration, researchers have attempted to shift fatty acid metabolism towards glycolysis, with mitochondria playing a crucial role in this process. The Randle cycle regulates fatty acid oxidation, where acetyl-CoA generated by mitochondria inhibits pyruvate dehydrogenase (PDH), thereby suppressing the impact of glycolysis on myocardial cell metabolism [49]. Feeding neonatal mice with milk lacking fatty acids can extend the window of myocardial cell proliferation after birth, while conditional knockout mice of pyruvate dehydrogenase kinase (PDK) 4 show reduced DNA damage and myocardial cell cycle arrest DNA damage response (DDR) markers, promoting myocardial cell proliferation [12]. Carnitine O-palmitoyltransferase (CPT) 1 is a key enzyme regulating mitochondrial fatty acid uptake [50]. When endogenous levels of malonyl-CoA increase, CPT1 is inhibited, halting mitochondrial fatty acid uptake, and malonyl-CoA decarboxylase (MCD) participates in the decarboxylation of malonyl-CoA to acetyl-CoA, thus inhibiting MCD can reduce fatty acid oxidation to protect ischemic myocardium [51]. Li et al. used inactivated CPT1b to inhibit the fatty acid oxidation pathway and improve myocardial cell survival and proliferation [17]. Furthermore, recent studies suggest that simple alterations in myocardial metabolic substrates may not necessarily be the optimal strategy for promoting repair and regeneration after infarction [18]. A deeper understanding of the regulation mechanisms of mitochondrial fatty acid transport and β-oxidation pathways is crucial for developing new therapeutic approaches (Fig. 1).

The principal regulatory nodes for the oxidation of fatty acids and glucose-derived pyruvate in mitochondria are carnitine palmitoyltransferase 1 (CPT1) and pyruvate dehydrogenase complex (PDC), respectively. CPT1 facilitates the translocation of fatty acids into mitochondria, enabling β-oxidation. The coactivator PGC-1α is modulated through the concerted actions of AMP-activated protein kinase (AMPK) and Sirtuin 1 (SIRT1). AMPK, a serine/threonine kinase, is activated in response to elevated AMP/ATP ratios, leading to the phosphorylation of several transcription factors, including nuclear respiratory factor 1 (NRF-1), cAMP response element-binding protein (CREB), and myocyte enhancer factor 2 (MEF2). This kinase also targets metabolic enzymes for phosphorylation. The interconnection between the mitochondrial lifecycle and PGC-1α is underscored by mitochondrial biogenesis, which is precipitated by an energy deficit perceived by AMPK. This process is characterized by the augmented activity or expression of mitochondrial transcription factor A (TFAM), which localizes to mitochondria, associates with mitochondrial DNA (mtDNA), and stimulates its transcription and duplication. Hypoxia-inducible factor 1 (HIF-1) is upregulated by low oxygen levels and upregulates pyruvate dehydrogenase kinase 1 (PDK1), curtailing excessive mitochondrial reactive oxygen species (ROS) production by attenuating mitochondrial respiration
More and more research reports indicate a close association between circRNA and oxidative stress [52]. Circular RNA (circRNA) is a type of covalently closed RNA sequence lacking a 5'-cap and 3'-polyadenylated tail, with most of them lacking protein-coding ability. Due to their circular structure, circRNA is more stable compared to linear RNA [53]. Several circRNAs, such as circNfix [11], circFndc3b [54], and circHipk3 [13], have been reported to play crucial roles in cardiac regeneration and repair. CircRNAs can regulate gene expression associated with ROS production by serving as miRNA sponges, thereby influencing cellular oxidative stress responses and apoptotic pathways [55]. Additionally, the biosynthesis, localization, and degradation processes of circRNAs are precisely regulated, which helps maintain intracellular ROS balance and cope with oxidative stress.
Recent studies have revealed that certain circRNAs localize to mitochondria and participate in regulating the mitochondrial transcriptome, which has potential implications in maintaining intracellular ROS balance and regulating cell death and survival [56]. Vcp is expressed in various subcellular structures and cell types, with biological functions including involvement in protein degradation and homeostasis, assembly of organelle membranes, cell proliferation, and apoptosis [57]. Zheng et al. found that overexpression of mitochondrial-localized circSamd4 could control the opening of the mitochondrial permeability transition pore (mPTP) and the production of ROS within mitochondria by regulating Vcp protein translocation to mitochondria, thereby improving myocardial dysfunction induced by MI [15]. Ma et al. observed that the expression of circMdc1 was suppressed under oxidative stress conditions, revealing a potential link between circMdc1 and mitochondrial ROS production. Furthermore, circMdc1 mitigates DNA damage and promotes cardiomyocyte re-entry into the cell cycle by affecting the expression of its host gene, Mdc1 [16].
Epigenetic Regulation
Epigenetics studies changes in gene expression that do not involve alterations in the DNA sequence itself, but rather are achieved through mechanisms such as DNA methylation, histone modifications, chromatin accessibility adjustments, and RNA-mediated gene modifications. Epigenetic regulation affects myocardial regeneration by directly or indirectly influencing the transcriptional networks related to mitochondrial function within cardiac myocytes.
Uncoupling Protein (UCP) 2 can regulate myocardial cell cycle activity, acetyl-CoA, and histone acetylation, enabling cells to adapt to moderate hypoxia [21]. In mutated CPT1b cardiomyocytes, energy metabolism and activation of α-ketoglutarate-dependent lysine demethylase (KDM5) result in an increase in H3K4me3 methylation sites within mature gene loci, thereby reducing gene transcriptional activity and promoting the transition of cardiomyocytes to a less mature state, facilitating proliferation [17].
In Chen et al.'s study, experimental results demonstrate that both in vivo and in vitro, mitochondrial transmembrane protein (TMEM) 11 inhibits myocardial cell proliferation and regeneration. This effect involves the interaction between TMEM11 and methyltransferase-like protein (METTL) 1, which enhances m7G methylation of ATF5 mRNA, leading to increased ATF5 expression. ATF5, in a TMEM11-dependent manner, increases the transcription of lnca1, which is a cell cycle protein-dependent kinase inhibitor interacting with cyclin A1. Therefore, TMEM11-mediated m7G methylation is involved in the regulation of myocardial cell proliferation, and targeting the TMEM11-METTL1-ATF5-INCA1 axis could serve as a novel therapeutic strategy to promote cardiac repair and regeneration [22].
Silent mating type information regulation 2 homolog (SIRT) 1, a crucial NAD+-dependent deacetylase, influences various substrates including transcription factors and histones through its deacetylation activity, thereby directly regulating the metabolic state and stress resistance of myocardial cells. Specifically, the interaction between SIRT1 and peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α enhances PGC-1α activity through deacetylation, promoting the transcription of oxidative phosphorylation genes, increasing myocardial cell oxygen consumption rate, and energy production. In response to oxidative stress, the role of SIRT1 also reflects the effects of epigenetic regulation. SIRT1 activates antioxidant enzymes such as manganese-dependent superoxide dismutase (MnSOD) and catalase by deacetylating FOXO1/3, thereby alleviating oxidative stress in mitochondria and cells [58]. Under conditions of heart failure, the Ataxia Telangiectasia Mutated (ATM)-mediated DNA damage response pathway is activated, leading to the ubiquitination and degradation of LARP7, which subsequently affects the stability and deacetylase activity of SIRT1, impacting mitochondrial biogenesis and oxidative phosphorylation capacity in myocardial cells [20].
MicroRNAs (miRNAs) are non-coding single-stranded RNAs that regulate gene expression and mRNA silencing, participating in cardiac development and myocardial cell proliferation processes [59]. For example, the miR-302-367 cluster promotes myocardial cell mitosis by targeting the Hippo pathway or upregulating the expression of the homeobox transcription factor Pitx2. Pitx2 can also activate the expression of ROS scavengers, Nrf2, and electron transport chain genes, which are closely related to mitochondrial function [60]. Additionally, the regulatory role of long non-coding RNAs (lncRNAs) plays an important role in cardiac repair after myocardial infarction. Bioinformatics analysis, luciferase reporter experiments, and RNA pull-down assays have revealed that Malat1 acts as a competitive endogenous RNA (ceRNA) for miR-26b-5p, forming a signaling axis with Mfn1 to regulate mitochondrial dynamics and endothelial function. Overexpression of Mfn1 can significantly reverse microvascular dysfunction and cardiac microvascular endothelial cell (CMEC) damage caused by Malat1 silencing, primarily by reducing mitochondrial fragmentation and inhibiting mitochondria-dependent cell apoptosis [19].
Signaling Between Mitochondria and the Cell Nucleus
In cardiomyocytes, the signaling between the nucleus and mitochondria is orchestrated through intricate mechanisms and signaling pathways, enabling these two organelles to mutually regulate their functions to meet cellular demands. Nuclear-encoded proteins are responsible for regulating mitochondrial function, while mitochondria, when functionally impaired or energy production is inadequate, signal to the nucleus through a series of signaling mechanisms to adjust nuclear gene expression in response to energy demands and cellular stress [61].
Peroxisome proliferator-activated receptor-alpha (PPARα) is highly expressed in the heart and serves as a nuclear receptor responsible for transporting fatty acids into mitochondria and peroxisomes [62]. PPARα can alter myocardial metabolism patterns by forming heterodimers with other ligands such as retinoid X receptor (RXR), binding to specific elements in the promoter regions of its target genes. Transcript factor sequence analysis has revealed an enrichment of DR1 motifs at RXR target gene sites, including those associated with mitochondrial fatty acid oxidation (mtFAO) [26]. PGC-1α/β are important co-regulators of PPARα, participating in the expression of multiple genes related to mitochondrial respiration and functioning in fatty acid beta-oxidation [23, 63]. Moreover, high expression of PGC-1 in cardiomyocytes induces mitochondrial biogenesis and oxidative capacity, while low ATP levels trigger AMP-activated protein kinase (AMPK) to phosphorylate PGC-1α, leading to its nuclear translocation and upregulation of nuclear respiratory factor (Nrf)-1 and mitochondrial transcription Factor A (TFAM) to coordinate mitochondrial biogenesis [64]. Estrogen-related receptors (ERRs), belonging to ligand-independent nuclear receptors, interact with PGC-1α/β to collectively activate downstream transcriptional events [65]. Studies by Sakamono et al. have further demonstrated the necessity of PGC-1 effectors ERRα/γ in properly controlling cardiac myocyte differentiation gene switches and normal cardiac maturation [24].
The relationship between AMPK and mitochondria primarily manifests in two aspects: energy sensing transduction and regulation of mitochondrial function. Activation of AMPK can promote mitochondrial biogenesis and dynamics to increase mitochondrial energy production capacity [25, 27]. AMPK also regulates metabolic pathways within mitochondria, such as promoting glucose uptake, activating glycolysis, inhibiting glycogen synthesis, activating fatty acid oxidation, and inhibiting lipid synthesis, thereby increasing fatty acid breakdown to generate energy [66]. AMPK is a critical regulatory factor in cellular energy sensing and maintaining energy homeostasis, with its activation regulated by changes in ATP/AMP concentration ratios. When energy utilization changes, the ratios of ATP/ADP or ATP/AMP also fluctuate, leading to phosphorylation and activation of AMPK by key proteins in various pathways, including the mTORC1 complex, lipid homeostasis, glycolysis, and mitochondrial homeostasis [67].
A recent study has revealed extensive reshaping of sphingolipid metabolism in neonatal heart injury, particularly emphasizing the crucial role of sphingosine-1-phosphate (S1P) generation in cardiac regeneration. The study indicates that two isoforms of sphingosine kinases, SphK1 and SphK2, regulate cardiac regeneration through different mechanisms: SphK2 is downregulated during cardiac development, and its activation induces re-entry into the cell cycle and cytokinesis in adult CM, promoting regeneration; whereas SphK1 promotes fibrotic autocrine mechanisms in cardiac fibroblasts [68].
Mitochondrial retrograde signaling can be achieved through various mechanisms, including direct modulation of transcription factor activity, alteration of intracellular metabolite levels (such as ATP, ROS, αKG, etc.), and responses mediated by specific signaling molecules. For instance, interruption of mitochondrial protein translation and fatty acid oxidation metabolism regulates specific gene expression in cardiomyocytes to promote cell proliferation through two different intermediates: the mitochondrial unfolded protein response (UPRmt) mediated by activating transcription factor (ATF)4 and H3K4me3 modification induced by αKG and Kdm5 [69].
Translational Medicine Advances in Cardiac Regeneration Research
The translational medical progress in cardiac regeneration research has advanced from basic science laboratories, showing potential prospects for treating heart diseases. Mitochondria play crucial roles in coordinating energy metabolism, oxidative stress levels, and signaling pathways in cardiac regeneration.
In cardiac cells, excessive ROS can lead to DNA damage and cell cycle arrest. Therefore, researchers have developed various novel ROS scavengers. For example, Zhang et al. constructed emerging hybrid cell-derived extracellular vesicles containing a NADPH analog—MitoN—targeting mitochondria to eliminate ROS accumulation-induced cell cycle arrest [70]. Xiang et al. proposed a dual-metal nanoenzyme (Cu-TCPP-Mn) that mimics the functions of superoxide dismutase (SOD) and catalase, effectively scavenging ROS and alleviating inflammation with excellent enzyme activity. Experimental verification in animal models of MI and myocardial ischemia-reperfusion (I/R) injury demonstrated that this dual-metal nanoenzyme could protect cardiac tissues from oxidative stress and inflammation damage, aiding in cardiac function recovery [71]. Wang et al. developed a novel on-demand adaptive drug-releasing hydrogelthat clears excess ROS and releases 1,4-dihydroxybenzene-4-keto-3-carboxylic acid (DPCA) to modulate the inflammatory microenvironment and inhibit cardiac fibroblast proliferation after myocardial infarction, thereby maintaining HIF-1α expression [72]. Diao et al. investigated the effects of the commonly used Del Nido (DN) cardioplegic solution on the Nrf2 transcription factor during heart surgery. Transcriptomic analysis using RNA-seq and luciferase reporter gene assays confirmed the activation of Nrf2 and its impact on antioxidant gene expression. Additionally, high concentrations of potassium in DN solution significantly induced Nrf2 expression and activity in human cardiac cells. Therefore, using DN cardioplegic solution and potassium during surgery can induce Nrf2 expression and activity to protect the heart from oxidative damage [73].
miRNA has broad applications and can rapidly control gene expression. Over the past decade, researchers have identified specific miRNAs related to the heart, and miRNA-based therapies have benefited from inducing terminal differentiation, proliferation, and migration of cardiac myocytes. Reversing the transition of cardiac myocytes from mitotic arrest to proliferative progenitor cells can facilitate tissue repair and regeneration [59]. For example, MGN-1374 is an anti-miR drug targeting the miR-15 family. MiR-15 can regulate processes such as cardiac myocyte survival, proliferation, and death. By inhibiting the miR-15 family, MGN-1374 can alleviate the inhibition of cardiac myocyte proliferation, thereby promoting repair and regeneration of the heart after myocardial infarction. Concurrently, MGN-1374 demonstrates the potential of using miRNA technology to target specific molecular targets for the treatment of heart disease [74, 75].
Chen et al. utilized CRISPR/Cas9 technology to generate mice with cardiac myocyte-specific deletion of the lactate dehydrogenase A(LDHA) gene. Metabolomics, proteomics, and Co-IP experiments indicated that LDHA-mediated reduction of succinyl-CoA inhibited succinylation ubiquitination dependent on thioredoxin reductase 1 (Txnrd1), thereby reducing ROS levels [76]. Additionally, a substantial body of recent research suggests that mitochondrial transplantation is an alternative therapy aimed at increasing mitochondrial quantity and improving mitochondrial function, offering significant therapeutic potential. Mitochondrial transplantation has been shown to significantly improve the function and prognosis of cardiovascular disease patients [77]. Mitochondrial transplantation has been shown to promote the energy and mechanical contraction functions of cardiomyocytes, reduce cell apoptosis, macrophage infiltration, and inflammatory responses, thereby alleviating damage. Sun and colleagues combined a specific peptide (PEP) with triphenyl phosphonium cation (TPP+) to form a PEP-TPP-mitochondria compound, aimed at addressing the issue of mitochondrial transfer to ischemic myocardium [78].
Conclusion
Myocardial regeneration is a complex regulatory process with multiple factors and stages, in which mitochondria demonstrate significant potential in regulating myocardial regeneration during myocardial cell proliferation and differentiation. Under the backdrop of heart disease, the oxidative stress response of myocardial cells differs from the normal state. The dynamic changes of mitochondria not only affect mitochondrial energy generation capacity but may also impact myocardial cell signaling and gene expression, warranting deeper investigation into the molecular mechanisms, particularly how they affect myocardial cell proliferation and differentiation. Future research could focus more on the role of specific enzymes in mitochondrial metabolism, the oxidative stress response of myocardial cells under disease conditions, and the molecular mechanisms in mitochondrial dynamic changes. By modulating mitochondria, changing the energy metabolic state of myocardial cells, reducing oxidative stress, and creating a regenerative microenvironment, myocardial proliferation and regeneration can be promoted. This offers new directions for overcoming the current challenges faced by myocardial regeneration therapies.
From a translational medicine perspective, although progress has been made in the field of myocardial regeneration, there are still difficulties and challenges. For example, the inability of adult cells to generate new myocardial cells, clinical issues arising from pluripotent stem cell (PSC) therapy (such as arrhythmias, immune responses, etc.), and the bleak prospects of extracellular vesicle-induced myocardial proliferation [79]. These issues prompt a shift in research direction from promoting exogenous to enhancing endogenous myocardial regeneration, where the study of mitochondrial regulation mechanisms in myocardial regeneration holds greater translational potential. Future interventions could involve targeted regulation of signaling pathways, pharmacological improvement of metabolic states, and more secure, precise, and potentially personalized treatments for heart failure.
Data availability
The data used in this study is publicly available.
Abbreviations
- αKG:
-
α-ketoglutarate
- ATF:
-
Activating Transcription Factor
- ATM:
-
Ataxia Telangiectasia Mutated
- AMPK:
-
AMP-Activated Protein Kinase
- ceRNA:
-
Competitive Endogenous RNA
- CMEC:
-
Cardiac Microvascular Endothelial Cell
- CVD:
-
Cardiovascular Diseases
- CPT1:
-
Carnitine Palmitoyltransferase 1
- DAMPs:
-
Damage-Associated Molecular Patterns
- DDR:
-
DNA Damage Response
- DN:
-
Del Nido
- ERRs:
-
Estrogen-Related Receptors
- IR:
-
Ischemia-Reperfusion
- LDHA:
-
Lactate Dehydrogenase A
- lncRNA:
-
Long Non-Coding RNA
- MCD:
-
Malonyl-CoA Decarboxylase
- MI:
-
Myocardial Infarction
- miRNAs:
-
MicroRNAs
- Nrf:
-
Nuclear respiratory Factor
- PDH:
-
Pyruvate Dehydrogenase
- PDK4:
-
Pyruvate Dehydrogenase Kinase 4
- PGC:
-
Peroxisome Proliferator-Activated Receptor γ Coactivator
- ROS:
-
Reactive Oxygen Species
- TFAM:
-
Mitochondrial Transcription Factor A
- TMEM:
-
Transmembrane Protein
- UCP:
-
Uncoupling Protein
References
WHO. Cardiovascular diseases (CVDs). https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) 2021.
Wu X, Reboll MR, Korf-Klingebiel M, et al. Angiogenesis after acute myocardial infarction. Cardiovasc Res. 2021;117(5):1257–73.
Guo Q-Y, Yang J-Q, Feng X-X, et al. Regeneration of the heart: from molecular mechanisms to clinical therapeutics. Military Med Res. 2023;10(1):18.
Sayers JR, Riley PR. Heart regeneration: beyond new muscle and vessels. Cardiovasc Res. 2021;117(3):727–42.
Harrington J, Jones WS, Udell JA, et al. Acute Decompensated Heart Failure in the Setting of Acute Coronary Syndrome. JACC. Heart Failure. 2022;10(6):404–14.
Klaourakis K, Vieira JM, Riley PR. The evolving cardiac lymphatic vasculature in development, repair and regeneration. Nat Rev Cardiol. 2021;18(5):368–79.
Harrington J, Petrie MC, Anker SD, et al. Evaluating the Application of Chronic Heart Failure Therapies and Developing Treatments in Individuals With Recent Myocardial Infarction: A Review. JAMA Cardiol. 2022;7(10):1067–75.
Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324(5923):98–102.
Rigaud VOC, Hoy RC, Kurian J, et al. RNA-Binding Protein LIN28a Regulates New Myocyte Formation in the Heart Through Long Noncoding RNA-H19. Circulation. 2023;147(4):324–37.
Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010;56(2):130–40.
Huang S, Li X, Zheng H, et al. Loss of Super-Enhancer-Regulated circRNA Nfix Induces Cardiac Regeneration After Myocardial Infarction in Adult Mice. Circulation. 2019;139(25):2857–76.
Cardoso AC, Lam NT, Savla JJ, et al. Mitochondrial Substrate Utilization Regulates Cardiomyocyte Cell Cycle Progression. Nat Metab. 2020;2(2):167–78.
Si X, Zheng H, Wei G, et al. circRNA Hipk3 Induces Cardiac Regeneration after Myocardial Infarction in Mice by Binding to Notch1 and miR-133a. Mol Ther Nucleic Acids. 2020;21:636–55.
Fajardo VM, Feng I, Chen BY, et al. GLUT1 overexpression enhances glucose metabolism and promotes neonatal heart regeneration. Sci Rep. 2021;11(1):8669.
Zheng H, Huang S, Wei G, et al. CircRNA Samd4 induces cardiac repair after myocardial infarction by blocking mitochondria-derived ROS output. Mol Ther: J Am Soc Gene Ther. 2022;30(11):3477–98.
Ma W, Wang X, Sun H, et al. Oxidant stress-sensitive circRNA Mdc1 controls cardiomyocyte chromosome stability and cell cycle re-entry during heart regeneration. Pharmacol Res. 2022;184:106422.
Li X, Wu F, Günther S, et al. Inhibition of fatty acid oxidation enables heart regeneration in adult mice. Nature. 2023;622(7983):619–26.
Kankuri E, Finckenberg P, Leinonen J, et al. Altered acylcarnitine metabolism and inflexible mitochondrial fuel utilization characterize the loss of neonatal myocardial regeneration capacity. Exp Mol Med. 2023;55(4):806–17.
Chen Y, Li S, Zhang Y, et al. The lncRNA Malat1 regulates microvascular function after myocardial infarction in mice via miR-26b-5p/Mfn1 axis-mediated mitochondrial dynamics. Redox Biol. 2021;41:101910.
Yu H, Zhang F, Yan P, et al. LARP7 Protects Against Heart Failure by Enhancing Mitochondrial Biogenesis. Circulation. 2021;143(20):2007–22.
Rigaud VO, Zarka C, Kurian J, et al. UCP2 modulates cardiomyocyte cell cycle activity, acetyl-CoA, and histone acetylation in response to moderate hypoxia. JCI Insight. 2022;7(15)
Chen X-Z, Li X-M, Xu S-J, et al. TMEM11 regulates cardiomyocyte proliferation and cardiac repair via METTL1-mediated m(7)G methylation of ATF5 mRNA. Cell Death Differ. 2023;30(7):1786–98.
Northam C, LeMoine CMR. Metabolic regulation by the PGC-1α and PGC-1β coactivators in larval zebrafish (Danio rerio). Comparative biochemistry and physiology. Part A, Mol Integ Physiol. 2019;234:60–7.
Sakamoto T, Matsuura TR, Wan S, et al. A Critical Role for Estrogen-Related Receptor Signaling in Cardiac Maturation. Circ Res. 2020;126(12):1685–702.
Hu Y, Chen H, Zhang L, et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy. 2021;17(5):1142–56.
Paredes A, Justo-Méndez R, Jiménez-Blasco D, et al. γ-Linolenic acid in maternal milk drives cardiac metabolic maturation. Nature. 2023;618(7964):365–73.
Malik N, Ferreira BI, Hollstein PE, et al. Induction of lysosomal and mitochondrial biogenesis by AMPK phosphorylation of FNIP1. Science. 2023;380(6642):eabj5559.
Ramachandra CJA, Chua J, Cong S, et al. Human-induced pluripotent stem cells for modelling metabolic perturbations and impaired bioenergetics underlying cardiomyopathies. Cardiovasc Res. 2021;117(3):694–711.
Brown DA, Perry JB, Allen ME, et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14(4):238–50.
Picard M, Shirihai OS. Mitochondrial signal transduction. Cell Metab. 2022;34(11):1620–53.
Mejia EM, Hatch GM. Mitochondrial phospholipids: role in mitochondrial function. J Bioenerg Biomembr. 2016;48(2):99–112.
Giacomello M, Pyakurel A, Glytsou C, et al. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21(4):204–24.
Vringer E, Tait SWG. Mitochondria and cell death-associated inflammation. Cell Death Different. 2023;30(2):304–12.
Schiaffarino O, Valdivieso González D, García-Pérez IM, et al. Mitochondrial membrane models built from native lipid extracts: Interfacial and transport properties. Front Mol Biosci. 2022;9:910936.
Vogel F, Bornhövd C, Neupert W, et al. Dynamic subcompartmentalization of the mitochondrial inner membrane. The J Cell Biol. 2006;175(2):237–47.
Yan C, Duanmu X, Zeng L, et al. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells. 2019;8(4)
Quintana-Cabrera R, Scorrano L. Determinants and outcomes of mitochondrial dynamics. Mol Cell. 2023;83(6):857–76.
Lu Y, Li Z, Zhang S, et al. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. 2023;13(2):736–66.
Ajoolabady A, Chiong M, Lavandero S, et al. Mitophagy in cardiovascular diseases: molecular mechanisms, pathogenesis, and treatment. Trends Mol Med. 2022;28(10):836–49.
Yu W, Sun S, Xu H, et al. TBC1D15/RAB7-regulated mitochondria-lysosome interaction confers cardioprotection against acute myocardial infarction-induced cardiac injury. Theranostics. 2020;10(24):11244–63.
Sakaguchi A, Kimura W. Metabolic regulation of cardiac regeneration: roles of hypoxia, energy homeostasis, and mitochondrial dynamics. Curr Opinion Genetics Dev. 2021;70:54–60.
Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. The Int J Biochem Cell Biol. 2007;39(1):44–84.
Dai D-F, Chen T, Wanagat J, et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9(4):536–44.
Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Ann Rev Pharmacol Toxicol. 2007;47:143–83.
Chen QM. Nrf2 for protection against oxidant generation and mitochondrial damage in cardiac injury. Free Radical Biol Med. 2022;179:133–43.
Puente BN, Kimura W, Muralidhar SA, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157(3):565–79.
Honkoop H, de Bakker DE, Aharonov A, et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. eLife. 2019;8:e50163.
Notari M, Ventura-Rubio A, Bedford-Guaus SJ, et al. The local microenvironment limits the regenerative potential of the mouse neonatal heart. Sci Adv. 2018;4(5):eaao5553.
Rindler PM, Crewe CL, Fernandes J, et al. Redox regulation of insulin sensitivity due to enhanced fatty acid utilization in the mitochondria. Am J Physiol Heart Circ Physiol. 2013;305(5):H634–43.
McGarry JD, Takabayashi Y, Foster DW. The role of malonyl-coa in the coordination of fatty acid synthesis and oxidation in isolated rat hepatocytes. The J Biol Chem. 1978;253(22):8294–300.
Dyck JRB, Cheng J-F, Stanley WC, et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res. 2004;94(9):e78–84.
Saaoud F, Drummer IVC, Shao Y, et al. Circular RNAs are a novel type of non-coding RNAs in ROS regulation, cardiovascular metabolic inflammations and cancers. Pharmacol Therapeut. 2021;220:107715.
Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–57.
Garikipati VNS, Verma SK, Cheng Z, et al. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat Commun. 2019;10(1):4317.
Zhang Y, Chen Y, Wan Y, et al. Circular RNAs in the Regulation of Oxidative Stress. Front Pharmacol. 2021;12:697903.
Zhou W-Y, Cai Z-R, Liu J, et al. Circular RNA: metabolism, functions and interactions with proteins. Mol Cancer. 2020;19(1):172.
Shoshan-Barmatz V, Nahon-Crystal E, Shteinfer-Kuzmine A, et al. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol Res. 2018;131:87–101.
Rodgers JT, Lerin C, Haas W, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434(7029):113–8.
Shah V, Shah J. Restoring Ravaged Heart: Molecular Mechanisms and Clinical Application of miRNA in Heart Regeneration, vol. 9; 2022. p. 835138.
Tao G, Kahr PC, Morikawa Y, et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534(7605):119–23.
Fuchs P, Bohle F, Lichtenauer S, et al. Reductive stress triggers ANAC017-mediated retrograde signaling to safeguard the endoplasmic reticulum by boosting mitochondrial respiratory capacity. The Plant Cell. 2022;34(4):1375–95.
Goikoetxea MJ, Beaumont J, González A, et al. Altered cardiac expression of peroxisome proliferator-activated receptor-isoforms in patients with hypertensive heart disease. Cardiovasc Res. 2006;69(4):899–907.
Lin C-Y, Gurlo T, Haataja L, et al. Activation of peroxisome proliferator-activated receptor-gamma by rosiglitazone protects human islet cells against human islet amyloid polypeptide toxicity by a phosphatidylinositol 3’-kinase-dependent pathway. The J Clin Endocrinol Metab. 2005;90(12):6678–86.
Pandol SJ, Gottlieb RA. Calcium, mitochondria and the initiation of acute pancreatitis. Pancreatology. 2022;22(7):838–45.
Huss JM, Kopp RP, Kelly DP. Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. The J Biol Chem. 2002;277(43):40265–74.
Chun Y, Kim J. AMPK-mTOR Signaling and Cellular Adaptations in Hypoxia. Int J Mol Sci. 2021;22(18):9765.
Carling D. AMPK signalling in health and disease. Curr Opinion Cell Biol. 2017;45:31–7.
Ji X, Chen Z, Wang Q, et al. Sphingolipid metabolism controls mammalian heart regeneration. Cell Metab. 2024;S1550-4131(24):00017–2.
Li X, Zhu Y, Ruiz-Lozano P, et al. Mitochondrial-to-nuclear communications through multiple routes regulate cardiomyocyte proliferation. Cell Regeneration. 2024;13(1):2.
Zhang N, Fan M, Zhao Y, et al. Biomimetic and NOS-Responsive Nanomotor Deeply Delivery a Combination of MSC-EV and Mitochondrial ROS Scavenger and Promote Heart Repair and Regeneration. Adv Sci. 2023;10(21):e2301440.
Xiang K, Wu H, Liu Y, et al. MOF-derived bimetallic nanozyme to catalyze ROS scavenging for protection of myocardial injury. Theranostics. 2023;13(8):2721–33.
Wang K, Yao S-Y, Wang Z, et al. A Sequential Dual Functional Supramolecular Hydrogel with Promoted Drug Release to Scavenge ROS and Stabilize HIF-1α for Myocardial Infarction Treatment. Adv Healthcare Mater. 2024;13(6):e2302940.
Diao H, Dai W, Wurm D, et al. Del Nido cardioplegia or potassium induces Nrf2 and protects cardiomyocytes against oxidative stress. Am J Physiol Cell Physiol. 2023;325(6):C1401–14.
Chakraborty C, Sharma AR, Sharma G, et al. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol Ther Nucleic Acids. 2017;8:132–43.
Hydbring P, Badalian-Very G. Clinical applications of microRNAs [version 1; peer review: 2 approved]. F1000Res. 2013;2(136)
Chen Y, Wu G, Li M, et al. LDHA-mediated metabolic reprogramming promoted cardiomyocyte proliferation by alleviating ROS and inducing M2 macrophage polarization. Redox Biol. 2022;56:102446.
Sun M, Jiang W, Mu N, et al. Mitochondrial transplantation as a novel therapeutic strategy for cardiovascular diseases. J Transl Med. 2023;21(1):347.
Sun X, Chen H, Gao R, et al. Intravenous Transplantation of an Ischemic-specific Peptide-TPP-mitochondrial Compound Alleviates Myocardial Ischemic Reperfusion Injury. ACS Nano. 2023;17(2):896–909.
Zhang J, Bolli R, Garry DJ, et al. Basic and Translational Research in Cardiac Repair and Regeneration: JACC State-of-the-Art Review. J Am Coll Cardiol. 2021;78(21):2092–105.
Funding
This work was supported by grants from the Key Clinical Frontier Technology Project of Department of Science and Technology of Jiangsu Provincial (NO. BE2022806), and the National Natural Science Foundation of China (NO. 82370344).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
All authors have given consent for publication.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Associate Editor Nicola Smart oversaw the review of this article
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, YX., Zhao, AR., Wei, TW. et al. Progress of Mitochondrial Function Regulation in Cardiac Regeneration. J. of Cardiovasc. Trans. Res. (2024). https://doi.org/10.1007/s12265-024-10514-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12265-024-10514-w