
Abstract
The functional role of TGFβ type I receptor, activin-like kinase (ALK)-1 in post-myocardial infarction (MI) cardiac remodeling is unknown. We hypothesize that reduced ALK1 activity reduces survival and promotes cardiac fibrosis after MI. MI was induced in wild-type (WT), and ALK+/− mice by left coronary ligation. After 14 days ALK1+/− mice had reduced survival with a higher rate of cardiac rupture compared to WT mice. ALK1+/− left ventricles (LVs) had increased volumes at the end of systole and at the end of diastole. After MI ALK1+/− LVs had increased profibrotic SMAD3 signaling, type 1 collagen, and fibrosis as well as increased levels of TGFβ1 co-receptor, endoglin, VEGF, and ALK1 ligands BMP9 and BMP10. ALK1+/− LVs had decreased levels of stromal-derived factor 1α. These data identify the critical role of ALK1 in post-MI survival and cardiac remodeling and implicate ALK1 as a potential therapeutic target to improve survival after MI.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myocardial infarction (AMI) is a leading cause of heart failure and death [1, 2]. Cardiac fibrosis is a central component of cardiac remodeling after AMI. During the initial stages of the myocardial response to injury, cardiac fibrosis stabilizes the infarct zone and contributes to scar formation to myocardial rupture [3, 4]. However, excessive fibrosis may lead to maladaptive remodeling with impaired cardiac function and worsening tissue ischemia. The net result of maladaptive remodeling is worsening heart failure and LV dilatation [5]. Most therapeutic agents used to manage patients after AMI reduce the cardiac workload to limit maladaptive remodeling. Currently, no therapies that specifically target cardiac fibrosis in AMI or heart failure exist.
The TGFβ superfamily plays a major role in regulating cardiac fibrosis and post-AMI cardiac remodeling. TGFβ signaling plays a critical role in post-myocardial infarction wound healing by regulating the initial response to the infraction as well as regulating cardiac remodeling in the later stages [6,7,8]. Activin-like kinase 1 (ALK1) is an important receptor of the TGFβ superfamily and belongs to the type 1 receptor subtype that hetero-dimerizes with the type 2 receptor for signal transduction. Its role in regulating vascular homeostasis is well understood [9, 10]. The loss of its activity due to genetic mutations results in vascular malformation as seen in hereditary hemorrhagic telangiectasia type 2 (HHT2) [11].
ALK1 signaling is mediated through the SMAD1 pathway which attenuates cardiac fibrosis in murine models of heart failure [12]. Morine et al. have shown that partial loss of ALK1 is associated with increased fibrosis in the transverse aortic constriction (TAC)-mediated pressure overload model of heart failure [13]. We recently identified that the loss of BMP9, a high-affinity ligand of ALK1, is associated with higher cardiac fibrosis and maladaptive remodeling in TAC-induced heart failure in murine models [14] and further showed that the loss of BMP9 is associated with increased mortality and fibrosis in coronary artery ligation model of myocardial infarction. The role of ALK1 in post-infarct wound healing remains poorly understood. We sought to examine the role of ALK1 in post-myocardial infarction survival and cardiac remodeling.
Methods
Animals
All animal experiments were performed in accordance with the protocols approved by Tufts Medical Center IACUC. Wild-type (WT) mice were obtained from Jackson Laboratories. ALK1+/− mice were generously provided by Dr. Paul Oh.
Murine Model of Myocardial Infarction
Eight- to twelve-week-old mice were subjected to myocardial infarction by permanently ligating the left coronary artery (LCA). Initially, mice were anesthetized by isoflurane inhalation and put on mechanical ventilation using endotracheal intubation. A small incision was made between the ribs to expose the heart. The LCA was located and ligated using a suture to generate an infarct [15]. The wound was closed, and the mice were administered a single dose of buprenorphine SR for post-surgery recovery. Sham mice underwent the same process but without the LCA ligation. Mice were observed daily for survival for over 2 weeks.
Pressure Volume Measurements and Organ Harvesting
At the end of the study, all mice were subjected to a terminal procedure of pressure-volume (PV) measurement using a conductance catheter. For LV PV measurements, the carotid artery was located after making a small incision in anesthetized mice. A conductance catheter was inserted into the artery and secured using a suture. Then the catheter was advanced into the LV through the aortic valve. At the end of the recording, mice were euthanized, and tissues were harvested and immediately snap-frozen in liquid nitrogen and stored at −80 °C for further analysis. A section of the LV tissue was also stored in formalin for histology before snap-freezing in liquid nitrogen.
Protein Preparation for Western Blot
A small piece of snap-frozen tissue was dissected out and homogenized using pestles in T-Per lysis buffer supplemented with a protease inhibitor cocktail (PIC) and phosphatase inhibitor cocktail. The homogenate was incubated on ice for 1 h and then centrifuged at >13,000 RPM at 4 °C. The supernatant protein extract was separated and used for the preparation of western blot samples.
Western Blot
Protein samples were prepared using an equal amount of protein and were denatured by heating at 100 °C in Laemilii buffer (Boston Bioproducts, Milford, MA). Proteins were then separated on a mini-PROTEAN TGX 4–15% gel (BioRad, Hercules, CA) and transferred onto the PVDF membrane (MilliporeSigma, Burlington, MA). The membrane was blocked using a 5% solution of dry milk in PBS-T for 1 h. For the detection of specific proteins, membranes were incubated in specific antibodies overnight. The next day the membranes were washed three times for 10 min each with a 5% solution of dry milk in PBS-T. Membranes were then incubated with appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. In the end, membranes were washed three times with PBS-T for 10 min each. Protein bands were visualized using Pierce™ ECL Western Blotting Substrate (ThermoFisher Scientific, Waltham, MA) on a FluorChem E FE0504 imaging machine (ProteinSimple, San Jose, CA). ImageJ was used to quantify the band density. The following primary antibodies were used for the analysis. From Cell signaling: pSMAD1/5 (9516), SMAD1 (9743), SMAD3 (9513), pSMAD3 (9520), from Abcam: type 1 collagen (ab88147), from R and D systems: BMP9 (MAB3209), Endoglin (AF1320), MMP9 (AF909), from Novus: Vascular endothelial growth factor (VEGF) (NB100-664), from MilliporeSigma: GAPDH (MAB374).
Enzyme-Linked Immunosorbent Assay (ELISA)
Circulating levels of BMP9 and BMP10 were measured using ELISA kits from R and D systems: BMP9 (DY5566) and BMP10 (DY2926-05) using the manufacturer’s protocol. Serum samples collected at the time of tissue harvesting were used for these measurements. LV TGFb protein levels were measured using an ELISA kit (DB100B) from R and D systems.
Histology
Formalin-fixed samples were embedded in paraffin and subsequently sectioned. For Masson’s trichrome staining paraffin embedded heart tissue sections were deparaffinized with xylene and rehydrated in graded alcohol series. Sections were post-fixed in Bouin’s at 60 °C for 1 h. They were then stained with Weigert’s hematoxylin for 10 min and then washed in running water. Sections were then stained in Biebrich Scarlet Red for 15 min followed by differentiation using phosphotungstic-phosphomolybdic acid for 10 min. Sections were then stained with Aniline Blue for 15 min. Finally, sections were dehydrated in alcohol, cleared with xylene, and coverslipped with a nonaqueous mounting medium.
Measurement of Wall Thickness
The histological images were analyzed using ImageJ software to measure the cardiac wall thickness. The wall thickness was measured at three different locations in each remote region and infarct region of every section.
Statistical Analysis
In all graphs and the table, data are presented as mean with error bars representing standard deviation (SD). The Shapiro-Wilk test was used to confirm the normal distribution of the data set. For comparison among the groups, the Student’s T-test was used in the case of comparison between the two groups. For comparing more than two groups, analysis of variance (ANOVA) was used followed by Tukey’s post-hoc honest significant difference (HSD) test for multiple pairwise comparisons. For survival analysis, Kaplan-Meier curves were plotted and compared with the log-rank test. All statistical analysis was done using Prism GraphPad 9.3.1 software. A p-value of less than 0.05 was considered statistically significant.
Results
Partial Loss of ALK1 Reduces Survival After Myocardial Infarction and Increases LV Volume in Survivors
To understand the role of ALK1 in post-myocardial infarction healing, WT, and ALK1+/− mice were subjected to LAD ligation and observed for 14 days. ALK1+/− mice had significantly reduced survival as compared to WT mice (95.7% v 55%, p < 0.001). Further necropsy analysis revealed that most deaths in the ALK1+/− group occurred within 7 days of coronary ligation with more than 77% of these deaths involving cardiac rupture. No cardiac rupture was observed in WT mice after coronary ligation (Fig. 1a).

Partial loss of ALK1 loss leads to decreased survival and increased incidence of cardiac rupture and fibrosis. WT or ALK1+/− mice were subjected to LAD ligation to induce MI and were allowed to recover over 14 days to assess their survival. a Survival plot showing the percentage of mice that survived over 14 days after MI and incidences of cardiac ruptures among ALK1+/− deaths. ***p < 0.001, log-rank test, N = 20–23. b, c Heart sections with Masson’s trichrome staining showing MI with intact myocardium (WT MI) and ruptured myocardium (ALK1+/−) were observed at necropsy after MI, along with a graph of quantification of the fibrotic area as % of total LV area. **p < 0.01 Student’s t-test, N = 5–7
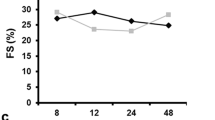
Among survivors, left ventricular (LV) end-diastolic volume (EDV) increased by 1.5-fold in ALK1+/− mice compared to WT (p = 0.018). LV end-systolic volumes (ESV) increased in ALK1+/− mice (p = 0.022). Both MI groups had decreased ejection fraction as compared to WT Sham animals (p = 0.005 for WT MI, p = 0.012 for ALK1+/− MI both vs WT Sham). Similar to EF, stroke work decreased significantly in both MI groups as compared to WT Sham animals (p = 0.002 for WT MI, p = 0.022 for ALK1+/− MI both vs WT Sham). The ESPVR remained unchanged among the groups while EDPVR increased significantly in the ALK1+/− MI group as compared to the WT sham group suggesting reduced LV compliance among ALK1+/− mice after MI (Table 1).
2 Weeks After MI, ALK+/− Mice Have Increased Fibrosis, Pro-fibrotic Signaling, and Collagen Deposition
To explore the underlying causes of increased mortality among ALK1+/− mice we quantified cardiac fibrosis and analyzed canonical TGFβ pro- and anti-fibrotic signaling. Compared to WT, histologic cardiac fibrosis increased to 26.6% in ALK+/− mice as compared to 13.6% in WT LVs, 2 weeks after MI (p = 0.006) (Fig. 1b, c). Measurement of cardiac wall thickness shows that while the infarct region has significantly reduced wall thickness as compared to the remote non-infarcted region, there is no significant difference in the wall thickness among the WT and ALK1+/− LVs in either region (Suppl. Fig. 1). The levels of pSMAD3 were more than 3-fold higher in LVs of ALK1+/− as compared to WT mice after MI (p = 0.039) (Fig. 2a, g). The levels of pSMAD1/5 decreased to 25% in ALK1+/− LVs as compared to WT LVs after MI (p = 0.003) (Fig. 2b, g). The LV levels of active TGFβ1 remained unchanged among all three groups (Supple Fig. 2). Partial loss of ALK1 also increased expression of α-SMA more than 3-fold in ALk1+/− LVs as compared to WT LVs (p = 0.002) (Fig. 2c, g), suggesting increased activation of fibroblasts. Type 1 collagen protein expression increased by 1.9-fold in ALK1+/− LVs as compared to WT Sham LVs (p = 0.021), while type 1 collagen mRNA levels were 7-fold higher in ALK1+/− LVs as compared to WT Sham (p=p=0.011) (Fig. 2d, e, g). Similar to type 1 collagen, periostin protein levels were increased 1.9-fold in ALK1+/− LVs as compared to WT Sham LVs (p = 0.023) (Fig. 2f, g). Taken together, these findings indicate that loss of ALK1 increases pro-fibrotic signaling and collagen deposition in ALK1+/− LVs after MI.

Partial ALK1 loss increased pro-fibrotic signaling, collagen, and periostin levels with increased expression of a marker of fibroblast activation in LVs, 2 weeks after MI. Western blot images and corresponding quantification graphs for relative protein expression of a pSMAD3, b pSMAD1/5 corrected for corresponding total SMADs, c α-SMA, d corrected for GAPDH. d, e Graphs showing mRNA and protein expression of type 1 collagen in LVs of mice 2 weeks after MI. f Graph showing relative protein levels of periostin corrected for GAPDH. *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA followed by Tukey’s HSD, N = 5–10
Loss of ALK1+/− Increased Expression of Angiogenic Markers and Decreased Expression of SDF1α in LVs After MI
LV levels of TGFβ co-receptor endoglin were 2.5-fold higher in ALK1+/− LVs as compared to WT LVs after MI (p < 0.0001). While endoglin protein levels were significantly altered, endoglin mRNA levels remained unchanged (Fig. 3a, d, i). LV protein levels of the ALK1 ligand BMP9 increased 5-fold in ALK1+/− LVs as compared to WT LVs after MI (p < 0.0001). Similar to BMP9 protein levels BMP9 mRNA levels significantly increased by more than 2-fold in ALK1+/− LVs as compared to WT LVs after MI (p = 0.039) (Fig. 2b, e, i). Another ALK1 ligand, BMP10, also showed a similar increase in protein as well as mRNA levels. BMP10 protein expression increased 2.6-fold in ALk1+/− LVs as compared to WT LVs after MI (p = 0.011). BMP10 mRNA expression increased more than 2-fold in ALK1+/− LVs as compared to WT LVs after MI (p = 0.013) (Fig. 2c, f, i). Surprisingly, the circulating levels of BMP9 and BMP10 increased significantly after MI in both WT and ALK1+/− animals. The basal circulating levels of both BMP9 and BMP10 were undetectable (Suppl. Fig. 3). Concomitantly ALK1 loss also increased VEGF expression in LVs by over 3-fold as compared to WT LVs after MI (p = 0.004) (Fig. 3g, i). SDF1α, a critical chemokine important for cell survival after MI, protein expression was reduced by 50% in ALK1+/− LVs as compared to WT LVs after MI (p = 0.0002) (Fig. 3h, i). These findings suggest that loss of ALK1 interferes with the angiogenesis process after MI (Fig. 4).

ALK1+/− mice have increased levels of proangiogenic proteins. Quantification graphs for protein and mRNA levels of a, d endoglin, b, e BMP9, and c, f BMP10 in LVs of mice from indicated groups 2 weeks after MI. Protein levels are corrected for GAPDH protein levels and mRNA levels are corrected for 18s expression level. g, h Quantification graphs for protein expression levels of G. VEGF and H. SDF1α corrected for GPDH protein levels. i Representative western blot images. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 one way ANOVA followed by Tukey’s HSD, N = 5–7

ALK1 is necessary for survival after MI and its partial loss is associated with increased incidences of myocardial rupture and death. Central figure depicting the mechanism of ALK1-mediated cardiac recovery process after MI. The intact BMP9/ALK1 axis preserves physiological scar formation while, partial loss of ALK1, increases fibrosis, and myocardial rupture along with increased expression of proangiogenic factors and decreased expression of SDF1α
Discussion
In this study, we report the critical role ALK1 plays in post-MI cardiac remodeling. Using a murine model of MI without reperfusion, we observed that partial loss of ALK1 in ALK1+/− mice severely reduced survival and increased cardiac fibrosis. Interestingly, despite having more fibrosis and collagen deposition, myocardium ruptured more frequently in ALK1+/− mice as compared to WT mice. Assessment of cardiac function revealed that ALK1+/− mice had enlarged ventricles. Analysis of molecular signaling in LVs 2 weeks after MI shows increased pro-fibrotic signaling mediated through SMAD3 while reduced anti-fibrotic SMAD1/5 signaling in ALK1+/− mice compared to WT. Further analysis of molecular changes in LVs revealed that ALK1+/− LVs have increased levels of BMP9 and BMP10, both high-affinity ligands of ALK1. Protein expression of TGFβ co-receptor endoglin was also significantly elevated in ALK1+/− LVs. Finally, we observed that VEGF levels were elevated and levels of SDF1α were significantly decreased in ALK1+/− LVs as compared to WT LVs. These data suggest that partial loss of ALK1 is detrimental to survival after MI and is associated with increased fibrosis.
The formation of a stable scar after MI is a critical step in post-MI cardiac remodeling. This process is tightly regulated by the TGFβ signaling pathway [16]. TGFβ-mediated SMAD3 phosphorylation activates pro-fibrotic processes that lead to increased collagen deposition and fibrosis [7]. SMAD1/5-mediated signaling mainly driven by BMP/ALK1 pathway is anti-fibrotic [14]. The balance between these two opposing signaling pathways is critical for the physiological response to myocardial injury and subsequent wound-healing process and stable scar formation after MI. Previous studies have shown that loss of BMP9, as well as ALK1, is associated with increased fibrosis and deteriorating cardiac function in pressure overload models of heart failure [13, 14]. Our earlier findings show that loss of BMP9 leads to increased incidences of cardiac rupture and reduced survival in the murine model of MI without reperfusion despite increased fibrosis [17]. Remarkably, in this report, we observed that partial loss of ALK1 leads to reduced survival and increased frequency of myocardial rupture after MI despite increased collagen deposition and fibrosis. These findings indicate that partial loss of signaling mediated through the BMP9/ALK1 signaling pathway is detrimental to post-MI survival. Interestingly, complete loss of BMP9 or partial loss of ALK1 does not affect the overall cardiac health during basal levels. Thus, suggesting the critical and specific role of the BMP9-ALK1 signaling pathway in post-MI wound healing. Partial loss of ALK1 also leads to an imbalance in SMAD3 and SMAD1/5 signaling with balance tipping in favor of pro-fibrotic SMAD3 signaling. Whether this imbalance leads to the weakening of the myocardium thus resulting in rupture remains to be investigated.
Previously, it has been shown that the collagen fiber alignment in post-MI scar formation is critical for the strength of the scar. Furthermore, post-MI wound healing changes the composition of ECM. Whether loss of BMP9/ALK1 signaling alters the composition of ECM and collagen alignment and thus compromises the structural integrity of the myocardium needs to be investigated [3, 18]. Furthermore, it is important to acknowledge that several other cellular pathways can contribute to the process of fibrosis and reorganization of ECM after MI. Inflammation and other immune system responses are known to be involved in the profibrotic process. However, the scope of this study was to understand the role of the ALK1/BMP9 signaling pathway in the larger context of TGFβ family signaling in response to wound healing after MI. Whether the ablation of ALK1 affects other cellular pathways that contribute to the process of fibrosis remains an active area of research.
The functional assessment of LV in the survivors 2 weeks after MI revealed that mice with reduced ALK1 levels had increased LV volumes at the end of systole as well as at the end of diastole. This indicates that the loss of ALK1 leads to maladaptive remodeling in LVs 2 weeks after MI. These findings are consistent with a previous report suggesting that partial loss of ALK1 is associated with the development of heart failure in the pressure overload model in mice [13]. While PV loop measurements using a catheter are precise in measuring actual pressure and volume relation, this method lacks the ability to assess the anatomical changes in the myocardium during the cardiac remodeling thus limiting the ability to measure structural components such as wall thickness. Thus, we utilized the histological images to measure the LV wall thickness in the infarct zone and remote non-infarcted region.
To further understand the underlying mechanism, we assessed the protein levels of proteins involved in the repair mechanism. Interestingly BMP9 and BMP10, both high-affinity ligands of ALK1 [19, 20] were significantly upregulated in ALK1+/− LVs after MI. This could be a part of the auto-regulatory process in which loss of ALK1 signaling leads to activation of signaling pathways that leads to increased expression of BMPs. Whether a switch in SMAD signaling plays a role in BMP9 and BMP10 regulation remains unclear. Angiogenesis is a major component of the post-MI wound healing process [21]. During the healing phase of myocardial wound healing after MI, new vessels are formed to restore the circulation in the infarcted zone. This process is also driven by hypoxia. ALK1-mediated signaling plays a critical role in angiogenesis. It inhibits the proliferation and migration of endothelial cells thus inhibiting angiogenesis while mediating the maturation of vessels [9]. Our data shows that angiogenic markers endoglin and VEGF protein expression were upregulated in ALK1+/− LVs compared to WT after MI. Increased expression of these factors also indicates increased hypoxia [22]. Shao et al. have previously shown that VEGF expression is regulated by ALK1-mediated signaling. siRNA-mediated loss of ALK1 is associated with increased expression of VEGF in endothelial cells. Furthermore, there is increased expression of VEGF in various organs of ALK1 deficient mice as compared to WT mice [23]. The increased expression of VEGF in ALK1+/− mice reported here is in line with these previous findings. The importance of ALK1 in angiogenesis is further underscored by the fact that mutations in the ALK1 gene and subsequent loss of its function are associated with type 2 HHT [11]. Type 2 HHT constitutes more than 50% of the entire population of HHT patients [24]. This functional loss of ALK1 is associated with arteriovenous malformations that lead to symptoms ranging from nosebleeds to bleeding in vital organs. In our mouse model of MI, we observe that ALK1+/− LVs have increased VEGF and endoglin suggesting increased angiogenesis, which is generally considered beneficial in post-MI healing. It is possible that loss of ALK1-mediated signaling leads to incomplete antigenic process leading to malformation of new vessels thus hampering the healing process and reduced survival. Further investigation is needed to determine the mechanisms by which ALK1 regulates the angiogenic process and how it impacts survival after MI.
Finally, we assessed the protein levels of an important chemokine, SDF1α. Several studies have shown that SDF1α and its receptor CXCR4-mediated signaling is a critical component of myocardial wound healing. It plays a critical role in the recruitment of progenitor cells to the site of injury and activates important pathways for cell survival thus limiting the infarct size [25, 26]. It also plays a critical role in angiogenesis [27, 28]. Previous studies have shown that TGFβ signaling regulates SDF1α expression in endothelial cells. BMP9 treatment increases SDF1α expression in HUVEC cells and this process is mediated through endoglin and ALK1 [29]. Interestingly, we observed that partial loss of ALK1 leads to decreased expression of SDF1α along with increased expression of endoglin and BMP9. This suggests that increased BMP9 and endoglin are not sufficient to upregulate SDF1α expression in the absence of complete ALK1 activity leading to deterioration of cardiac function and reduced survival.
Abbreviations
- MI:
-
Myocardial infarction
- TGFβ:
-
Transforming growth factor beta
- ALK1:
-
Activin-like kinase 1
- BMP:
-
Bone morphogenetic protein
- LCA:
-
Left coronary artery
- α-SMA:
-
α-smooth muscle actin
References
Kannel WB, Sorlie P, McNamara PM. Prognosis after initial myocardial infarction: the Framingham study. Am J Cardiol. 1979;44:53–9.
Virani SS, Alonso A, Benjamin EJ, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141:e139–596.
Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. 2016;365:563–81.
Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res. 2005;66:22–32.
Frantz S, Hundertmark MJ, Schulz-Menger J, Bengel FM, Bauersachs J. Left ventricular remodelling post-myocardial infarction: pathophysiology, imaging, and novel therapies. Eur Heart J. 2022;43(27):2549–61.
Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. CardiovascRes. 2007;74:184–95.
Bujak M, Ren G, Kweon HJ, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–38.
Matsui Y, Morimoto J, Uede T. Role of matricellular proteins in cardiac tissue remodeling after myocardial infarction. World J Biol Chem. 2010;1:69–80.
Lamouille S, Mallet C, Feige J-J, et al. Activin receptor–like kinase 1 is implicated in the maturation phase of angiogenesis. Blood. 2002;100:4495–501.
Oh SP, Seki T, Goss KA, et al. Activin receptor-like kinase 1 modulates transforming growth factor-β1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626–31.
Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–95.
Leask A. Getting to the heart of the matter. Circ Res. 2015;116:1269–76.
Morine KJ, Qiao X, Paruchuri V, et al. Reduced activin receptor-like kinase 1 activity promotes cardiac fibrosis in heart failure. CardiovascPathol. 2017;31:26–33.
Morine KJ, Qiao X, York S, et al. Bone morphogenetic protein 9 reduces cardiac fibrosis and improves cardiac function in heart failure. Circulation. 2018;138:513–26.
Fernández B, Durán AC, Fernández MC, et al. The coronary arteries of the C57BL/6 mouse strains: implications for comparison with mutant models. J Anat. 2008;212:12–8.
Scalise RFM, De Sarro R, Caracciolo A, et al. Fibrosis after myocardial infarction: an overview on cellular processes, molecular pathways, clinical evaluation and prognostic value. Med Sci. 2021;9:16.
Bhave S, Esposito M, Swain L, et al. Loss of bone morphogenetic protein-9 reduces survival and increases MMP activity after myocardial infarction. JACC Basic Transl Sci. 2023;8:1318–30.
Sullivan KE, Quinn KP, Tang KM, et al. Extracellular matrix remodeling following myocardial infarction influences the therapeutic potential of mesenchymal stem cells. Stem Cell Res Ther. 2014;5:14.
David L, Mallet C, Mazerbourg S, et al. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109:1953–61.
Scharpfenecker M, Floot B, Russell NS, et al. The TGF-Î2 co-receptor endoglin regulates macrophage infiltration and cytokine production in the irradiated mouse kidney. RadiotherOncol. 2012;105:313–20.
Wu X, Reboll MR, Korf-Klingebiel M, et al. Angiogenesis after acute myocardial infarction. Cardiovasc Res. 2021;117:1257–73.
Sánchez-Elsner T, Botella LM, Velasco B, et al. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-β pathways*. J Biol Chem. 2002;277:43799–808.
Shao ES, Lin L, Yao Y, et al. Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood. 2009;114:2197–206.
McDonald J, Bayrak-Toydemir P, DeMille D, et al. Curaçao diagnostic criteria for hereditary hemorrhagic telangiectasia is highly predictive of a pathogenic variant in ENG or ACVRL1 (HHT1 and HHT2). Genet Med. 2020;22:1201–5.
Hu X, Dai S, Wu W-J, et al. Stromal cell–derived factor-1α confers protection against myocardial ischemia/reperfusion injury. Circulation. 2007;116:654–63.
Saxena A, Fish JE, White MD, et al. Stromal cell-derived factor-1alpha is cardioprotective after myocardial infarction. Circulation. 2008;117:2224–31.
Hiasa K, Ishibashi M, Ohtani K, et al. Gene transfer of stromal cell-derived factor-1alpha enhances ischemic vasculogenesis and angiogenesis via vascular endothelial growth factor/endothelial nitric oxide synthase-related pathway: next-generation chemokine therapy for therapeutic neovascularization. Circulation. 2004;109:2454–61.
Yamaguchi J, Kusano KF, Masuo O, et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation. 2003;107:1322–8.
Young K, Conley B, Romero D, et al. BMP9 regulates endoglin-dependent chemokine responses in endothelial cells. Blood. 2012;120:4263–73.
Funding
This work was funded by the National Institute of Health grants R01HL133215 (NKK) and R01HL139785 (NKK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
Dr. Kapur receives institutional grant support and speaker/consulting honoraria from Abbott, Abiomed, Boston Scientific, LivaNova, Medtronic, MD Start, and Precardia.
Human and Animal Rights and Informed Consent
No human studies were carried out for the generation of data in this article.
Clinical Relevance
Post myocardial infarction wound healing process is crucial for the patient’s survival as well as the development of heart disease. The ALK1/BMP9 pathway is an emerging target for the development of novel therapies for both cancers and cardiovascular diseases. Data presented here sheds more light on the role of this pathway in the cardiac wound healing process thus identifying unique molecular targets for future treatments.
Additional information
Associate Editor Joost Sluijter oversaw the review of this article
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 96 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bhave, S., Swain, L., Qiao, X. et al. ALK1 Deficiency Impairs the Wound-Healing Process and Increases Mortality in Murine Model of Myocardial Infarction. J. of Cardiovasc. Trans. Res. 17, 496–504 (2024). https://doi.org/10.1007/s12265-023-10471-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-023-10471-w