
Abstract
Penehyclidine hydrochloride (PHC) is an anticholinergic drug with cardioprotective effects. Ferroptosis is closely related to myocardial ischaemia-reperfusion injury (MIRI). In the present study, MIRI was induced in rats by left anterior descending coronary artery ligation. PHC pretreatment increased haemodynamic parameters and histopathological damage and reduced myocardial infarction size in the MIRI model. PHC pretreatment also inhibited ferroptosis, which was characterized by the decreased levels of Fe2+, 4-hydroxynonenal and ACSL4, and increased levels of GPX4, GSH-Px and GST. In response to 6 h of oxygen-glucose deprivation and 18 h of reoxygenation, PHC pretreatment had the same effects on these factors in H9c2 cells and reduced lipid ROS levels. Furthermore, ACSL4 overexpression reversed the protective effects of PHC on H9c2 cells. These results indicated that PHC inhibited MIRI through ACSL4-mediated ferroptosis. This study demonstrated that PHC could inhibit ferroptosis in MIRI and the relationship among PHC, ACSL4, ferroptosis and MIRI.
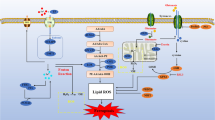
Graphical Abstract
This study demonstrated the inhibitory effect of PHC on ferroptosis and showed that PHC affects MIRI through ACSL4-mediated ferroptosis in vivo and in vitro.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocardial infarction is one of the main causes of death associated with cardiovascular diseases, which have high mortality rates worldwide. Early restoration of blood supply is the most important treatment for myocardial ischaemia. Reperfusion therapy after myocardial infarction, such as thrombolysis, percutaneous coronary intervention, and bypass surgery, can effectively improve blood supply [1]. However, when the blood supply is restored to the ischaemic myocardium, it can further damage energy metabolism, electrophysiology and tissue structure of the myocardium and heart function, and some of the damage may even be irreversible. This phenomenon is called myocardial ischaemia-reperfusion injury (MIRI) [2]. The pathogenesis of MIRI is complex, and involves oxygen free radicals, platelet activation, inflammatory mediators, and myocardial cell death [3, 4]. It is critical to clarify the molecular regulatory mechanism of MIRI so that it can be treated.
Ferroptosis, which was discovered in recent years, is a new form of cell death caused by an iron-dependent increase in lipid peroxides [5]. Ferroptosis cannot be regulated by inhibitors of apoptosis, pyroptosis and autophagy, but it can be effectively inhibited by lipophilic antioxidants and iron chelators [6]. The mechanism of ferroptosis is not well defined. Current studies mainly suggest that excessive iron ions in cells catalyse the production of hydroxyl free radicals through the Fenton reaction. Moreover, unsaturated fatty acids are further hydroxylated, which promotes the massive production of lipid peroxides and hydroperoxides. As a result, the structure and function of the cell membrane is severely damaged, causing an intracellular oxidative imbalance and iron-dependent lipid peroxidative death [7]. Acyl-CoA synthetase long-chain family member 4 (ACSL4) encodes a protein that is a key enzyme in fatty acid catabolism. ACSL4 is involved in the synthesis of oxidized membrane phospholipids and has been shown to be a key molecule that initiates ferroptosis in cells [8]. It was reported that ACSL4 knockout mitigated ferroptosis-mediated acute kidney injury in mice [9]. Ischaemia-induced ACSL4 activation exacerbates ferroptosis-mediated tissue damage during intestinal ischaemia-reperfusion [10]. Ferroptosis was induced in neurons during cerebral ischaemia-reperfusion by thrombin via ACSL4 activation [11]. Inhibiting ACSL4 attenuated ferroptosis after lung ischaemia-reperfusion injury [12]. Baicalin prevented MIRI by inhibiting ACSL4-mediated ferroptosis [13]. Another study reported that ferroptosis occurred in the reperfusion phase rather than the ischaemia phase, and the expression of ACSL4 increased with prolonged perfusion time [14]. This evidence suggests that inhibiting ACSL4-mediated ferroptosis is important for the treatment of MIRI.
Penehyclidine hydrochloride (PHC) is an anticholinergic drug with anti-muscarinic and anti-nicotinic activities [15]. PHC has been shown to reduce lung [16], kidney [17], small intestine [18], brain [19], and myocardial injury [20] caused by I/R. Possible mechanisms by which PHC reduces MIRI include the regulation of mitochondrial dynamics [21], inflammation [22], and apoptosis [23]. However, whether PHC reduces myocardial ischaemia-reperfusion injury by alleviating myocardial ferroptosis remains unclear. This study aimed to examine whether PHC is involved in MIRI through ACSL4-mediated ferroptosis.
Materials and Methods
Induction of MIRI
A total of 78 male Wistar rats weighing 250–300 g were kept in a temperature-controlled environment with a 12 h light/dark cycle and a relative humidity of 45–55%. Food and water were provided ad libitum. The rats were randomly divided into three groups: the Sham, MIRI and MIRI + PHC groups. Rats in the MIRI + PHC group were administered PHC (1 mg/kg) via tail vein injection. Rats in the Sham group and MIRI group were given the same amount of solvent in the same way. After 30 min, MIRI was induced by ligation of the left anterior descending coronary artery (LAD) for 45 min in the MIRI group and MIRI + PHC group. Rats in the Sham group were subjected to the same operation without ligation. After 3 h of reperfusion, a catheter was inserted from the right common carotid artery into the left ventricle. After the pressure sensor and the physiological signal collection system (BL420S, Chengdu Techman Instrument) were connected, haemodynamic parameters, including left ventricular end-diastolic pressure (LVEDP), left ventricular end-systolic pressure (LVESP) and the maximal ascending and descending rate of left ventricular pressure over time (± dp/dt max), were monitored. The haemodynamic data of rats in each group were collected, and then heart tissue samples were collected from the sacrificed rats. Three rats in the MIRI group and 2 rats in the PHC group died after surgery. One rat in the PHC group died during the haemodynamic examination. The animal experiments were performed in accordance with the ARRIVE guidelines and were approved by Capital Medical University (Ethics code: AEEI-2022-214).
Cell culture and Treatments
The rat cardiomyocyte H9c2 cell line was purchased from iCell Bioscience Inc and cultured in DMEM supplemented with 10% FBS at 37℃ with 5% CO2. Subsequent cell treatments were performed when the cells reached 90% confluence. H9c2 cells were first treated with 1, 2.5 or 5 μg/mL PHC for 4 h, and then cultured under oxygen-glucose deprivation and reoxygenation (OGD/R) conditions. In brief, the cells were incubated in glucose-free DMEM at 37℃ with 95% N2 and 5% CO2 for 6 h for OGD. Subsequently, the cells were cultured in normal medium for another 18 h culture under normal oxygen conditions. To assess whether ACSL4 is involved in PHC-mediated regulation of ferroptosis, H9c2 cells were transfected with a rat ACSL4 cDNA clone plasmid (G127995, YouBio) using Liposome 3000 reagent (Invitrogen). Briefly, the plasmid (2.5 μg) solution was mixed with diluted Liposome 3000 solution and then incubated at room temperature for 20 min. The transfection mixture was added dropwise to the cells. After 24 h of transfection, the cells were treated with PHC for 4 h, and cultured under OGD/R conditions.
Myocardial Histopathology
Haematoxylin‐eosin (HE) staining was used to histopathologically evaluate the heart. Following dehydration and clearing, the myocardial tissue was prepared into 5 μm paraffin sections, and dewaxed with anhydrous ethanol and xylene. The sections were soaked in distilled water for 2 min. Next, the sections were stained with haematoxylin (Solarbio) for 5 min and then stained with eosin (Sangon) for 3 min. Finally, staining was observed under a BX53 microscope.
Immunohistochemical (IHC) Evaluation of ACSL4 Expression
The tissue sections were placed into boiling antigen-repair solution and heated continuously at a low temperature for 10 min. To eliminate the activity of endogenous peroxidase, we incubated the slides with 3% hydrogen peroxide at room temperature for 15 min. Next, the sections were blocked with 1% BSA and then subjected to primary antibody incubation at 4℃ overnight. Secondary antibody incubation was performed at 37℃ for 60 min. The ACSL4 antibody (A6826, Abclonal) was diluted with PBS at a ratio of 1:50, and HRP-labelled goat anti-rabbit IgG (31460, Thermo Fisher) was diluted 1:500. After antibody incubation, DAB (Maxim) and haematoxylin (Solarbio) were used for colour development. Finally, the sections were dehydrated with graded ethanol, rendered transparent with xylene and mounted and sealed with neutral gum.
Immunofluorescence Staining of 4-hydroxynonenal
Paraffin-embedded myocardial tissue was cut into 5 μm sections and then treated with xylene and alcohol. After being washed 3 times with PBS, the tissue sections were treated with antigen retrieval solution for 10 min. The H9c2 cell slides were fixed with 4% paraformaldehyde for 15 min. PBS was used to wash away the paraformaldehyde. The cell slides were incubated in 0.1% Triton X-100 for 30 min and blocked with 1% bovine Serum albumin (BSA). Next, the tissue sections and cell slides were incubated with primary antibodies (4-hydroxynonenal, NBP2-59353, NOVUS) (1:50) at 4℃ overnight. The samples were incubated with secondary antibodies (Cy3-labeled goat anti-mouse IgG, A-21424, Invitrogen) (1:200) at room temperature for 60 min and stained with DAPI. Finally, staining was observed under a microscope.
Cell Viability Analysis
H9c2 cells were inoculated in 96-well culture plates at 5 × 103 cells per well. After PHC and OGD/R treatment, 10 μl of CCK-8 solution was added to each well and incubated for an additional 2 h. A microplate reader (800TS, Biotek) was used to measure the optical density at 450 nm.
Lipid Reactive Oxygen Species (ROS) Analysis
H9c2 cells were incubated with C11-BODIPY581/591 (Maokang Biotechnology) for 20 min. After being washed twice with PBS, the cells were subjected to flow cytometric analysis using a NovoCyte flow cytometer (Aceabio). The data were analysed with NovoExpress software (Aceabio).
Analysis of GSH-Px, GST and Ferrous Ions
We used a glutathione peroxidase (GSH-Px) assay kit (Nanjing Jiancheng), glutathione S-transferase (GST) activity assay kit (Solarbio) and ferrous iron colorimetric assay kit (Elabscience) to detect GSH-Px activity, GST activity and ferrous ion levels, respectively. Briefly, proteins were extracted from tissues or cells and subjected to concentration determination. GSH-Px activity was detected by enzymatic and chromogenic reactions. We used 0.1 g of myocardial tissue to detect ferrous ions, made a standard curve according to the kit instructions, and then measured the absorbance of the samples at 593 nm. To analyse GST activity, the absorbance at 340 nm was measured using a UV-vis spectrophotometer (Yoke Instrument), and enzyme activities were calculated according to the instructions provided by the kit manufacturer.
Western Blot (WB) Analysis
Cell lysis buffer for Western and IP (Beyotime) and PMSF were used for protein extraction. The protein concentration was estimated by a BCA Protein Assay Kit (Beyotime) and microplate reader (ELX-800, Biotek). Polyacrylamide gels were prepared consisting of 5% stacking gel, and 10% and 14% separating gel. The protein samples were diluted with 5 × loading buffer and PBS. The protein samples (20 μl) were separated by SDS-PAGE. The separated proteins were transferred to PVDF membranes (Millipore), which were blocked with 5% skim milk. Primary antibodies including ACSL4 (1:500, A6826, Abclonal), GPX4 (1:1000, A1933, Abclonal) and β-actin (1:1000, sc-47778, Santa Cruz) were added and incubated at 4℃ overnight, and secondary antibody incubation with HRP-labelled goat anti-rabbit IgG (1:5000, A0208, Beyotime) and HRP-labelled goat anti-mouse IgG (1:5000, A0216, Beyotime) was conducted at 37℃ for 45 min. A BeyoECL Plus kit (Beyotime) and Gel-Pro-Analyser were used to determine protein expression.
Real-time PCR
RNA extraction was performed using TRIpure reagent (BioTeke). RNA concentration was determined with a Nano 2000 spectrophotometer (Thermo Fisher). Reverse transcription of RNA into cDNA was conducted using BeyoRT™ II M-MLV reverse transcriptase (Beyotime) according to the manufacturer’s instructions. The reaction system was composed of 1 μl of cDNA, 0.5 μl of primer working solution (10 μM), 0.5 μl of SYBR Green (Solarbio), 10 μl of 2 × Taq PCR MasterMix (Solarbio) and 7.5 μl of ddH2O. The primer sequences for ACSL4 were as follows: sense sequence: 5’-TCCGCTTGTGACTTTAT-3’, anti-sense sequence: 5’-ACTTGGAGGAATGCTTG-3’. The program of the ExicyclerTM 96 Fluorometric quantitative instrument was as follows: 1) 95℃ for 5 min, 2) 95℃ for 10 s, 3) 60℃ for 10 s, 4) 72℃ for 15 s, 5) Go to step 2, Cycle 40, 6) 72℃ for 1 min 30 s, 7) 40℃ for 1 min, 8) Melting 60℃ to 94℃, 1℃/1 s, and 9) 25℃ for 1 min. The PCR data was analysed with the 2−△△CT method.
Results
PHC Treatment Alleviates Myocardial Injury Caused by Ischaemia Reperfusion
In rats with LAD ligation, PHC administration reduced LVEDP and increased LVESP and ± dp/dt max (Fig. 1a). To further evaluate the effects of PHC on myocardial injury, we performed TTC staining of myocardial tissues, and found that pretreatment with PHC decreased the infarction size (Fig. 1b). MIRI rats exhibited obvious myocardial histopathological damage, including increased inflammatory cell infiltration. As expected, myocardial histopathological damage was significantly reduced by PHC treatment (Fig. 1c). These results indicated that PHC pretreatment alleviated myocardial injury caused by ischaemia reperfusion in rats. In addition, IHC staining showed that PHC pretreatment inhibited the expression of ACSL4 in myocardial tissues (Fig. 1d).

PHC pretreatment alleviates myocardial injury caused by ischaemia-reperfusion. a Myocardial haemodynamics analysis of left ventricular end-diastolic pressure (LVEDP), left ventricular end-systolic pressure (LVESP), and the maximal ascending and descending rate of left ventricular pressure over time (± dp/dt max) in Wistar rats. b Myocardial infarction was detected by TTC staining and statistically analysed. c Myocardial histopathology was examined by HE staining; bar = 200 μm (100 ×) and 50 μm (400 ×). d The expression level of ACSL4 in myocardial tissues was detected by IHC staining; bar = 200 μm (100 ×) and 50 μm (400 ×). # p < 0.05, ## p < 0.01, ### p < 0.001, n = 6 in each group
PHC Inhibits Ferroptosis in Cardiomyocytes
Analysis of the relationship between PHC and the levels of ferroptosis-related factors showed that PHC reduced the level of ferrous iron (Fig. 2a) and increased the levels of GSH-Px (Fig. 2b) and GST (Fig. 2c) in cardiomyocytes. 4-Hydroxynonenal is a toxic by-product of lipid peroxidation. 4-Hydroxynonenal levels were analysed by immunofluorescence, and PHC suppressed the production of 4-hydroxynonenal (Fig. 2d). GPX4 is a ferroptosis suppressor, while ACSL4 is a ferroptosis promotor. The protein levels of GPX4 were increased by PHC pretreatment, while the protein levels of ACSL4 were decreased in rats treated with PHC (Fig. 2e). Therefore, we concluded that PHC inhibited ferroptosis in rat cardiomyocytes.

PHC inhibits ferroptosis in myocardial tissues. a Iron levels in myocardial tissues were measured by a ferrous iron colorimetric assay kit. ELISA was used to determine glutathione peroxidase (GSH-Px) b and glutathione S transferase (GST) c levels in myocardial tissues. d Immunofluorescence analysis of 4-hydroxynonenal in myocardial tissues; bar = 50 μm. e Protein levels of glutathione peroxidase 4 (GPX4) and acyl-CoA synthetase long-chain family member 4 (ACSL4) in myocardial tissues were determined by western blotting. # p < 0.05, ### p < 0.001, n = 6 in each group
PHC Attenuates OGD/R-induced Injury and Ferroptosis in H9c2 Cells
H9c2 cells were treated with 1, 2.5 or 5 μg/mL PHC for 4 h, and cultured under OGD/R conditions for 24 h. As a result, H9c2 cell viability improved in response to PHC treatment and showed a positive correlation with PHC concentrations (Fig. 3a). We selected 5 μg/mL PHC, which significantly improved cell viability, to evaluate its effects on ferroptosis in H9c2 cells. The levels of lipid ROS were assessed by flow cytometry, and statistical analysis showed that PHC reduced the production of lipid ROS (Fig. 3b). PHC pretreatment increased the protein levels of GST (Fig. 3c) and GSH-Px (Fig. 3d). The production of 4-hydroxynonenal and the protein levels of GPX4 and ACSL4 were measured in H9c2 cells, and PHC inhibited the production of 4-hydroxynonenal, decreased the level of ACSL4, and promoted the expression of GPX4 (Fig. 3e and f). These results were consistent with the in vivo results.

PHC attenuates OGD/R-induced injury and ferroptosis in H9c2 cells. a The viability of H9c2 cells treated with penehyclidine hydrochloride (PHC) under oxygen-glucose deprivation and reoxygenation (OGD/R) conditions. b Lipid ROS levels in H9c2 cells were assessed by BODIPY C11 staining coupled with flow cytometry. GST (c) and GSH-Px (d) levels in H9c2 cells were detected by ELISA. e Immunofluorescence analysis of 4-hydroxynonenal in H9c2 cells after PHC treatment; bar = 50 μm. (f). Protein levels of GPX4 and ACSL4 in H9c2 cells. ns: no significance, # p < 0.05, ## p < 0.01, ### p < 0.001, n = 3 in each group
PHC Alleviates OGD/R-induced H9c2 Cell Damage by Inhibiting ACSL4-Mediated Ferroptosis
Overexpression of ACSL4 was performed in H9c2 cells, and the efficiency was determined by WB analysis (Fig. 4a) and real-time PCR (Fig. 4b). H9c2 cells overexpressing ACSL4 were treated with 5 μg/mL PHC and cultured under OGD/R conditions. ACSL4 overexpression inhibited H9c2 cell viability (Fig. 4c). Interestingly, the production of 4-hydroxynonenal was increased in cells overexpressing ACSL4 (Fig. 4d). In addition, ACSL4 overexpression increased the level of lipid ROS (Fig. 4e) and decreased the levels of GST (Fig. 4f) and GSH-Px (Fig. 4g). The protein expression of GPX4 was inhibited by ACSL4 overexpression in H9c2 cells that were pretreated with PHC and subjected to OGD/R injury (Fig. 4h). These results revealed that ACSL4 overexpression reversed the effects of PHC on OGD/R injury.

PHC alleviates OGD/R-induced H9c2 cell damage by inhibiting ACSL4-mediated ferroptosis. Western blotting (a) and real-time PCR (b) verified the overexpression of ACSL4 in H9c2 cells. c The viability of H9c2 cells overexpressing ACSL4. d Immunofluorescence analysis of 4-hydroxynonenal in H9c2 cells overexpressing ACSL4; bar = 50 μm. e Lipid ROS levels in H9c2 cells overexpressing ACSL4. GST (f) and GSH-Px (g) levels in H9c2 cells overexpressing ACSL4. (h) Protein levels of GPX4 and ACSL4 in H9c2 cells overexpressing ACSL4. ns: no significance, # p < 0.05, ### p < 0.001, n = 3 in each group. OP: OGD/R + PHC
Discussion
During MIRI, PHC has been reported to regulate a variety of physiological processes except ferroptosis. Our study revealed that PHC alleviated myocardial reperfusion injury and improved cardiac function. We also found that PHC affected the levels of ferroptosis-related markers including Fe2+, ROS, GPX4, GSH-Px, 4-hydroxynonenal and ACSL4, and inhibited ferroptosis. This was the first time that PHC was shown to suppress ferroptosis in cardiomyocytes during MIRI.
In 2018, the Cell Death Nomenclature Committee reclassified cell death into two types: accidental cell death (ACD) and regulated cell death (RCD) [24]. ACD is uncontrolled. RCD which can be regulated, includes two forms. One is apoptotic, and the other is nonapoptotic. Nonapoptotic death includes autophagy, necroptosis, pyroptosis and ferroptosis [25]. Ferroptosis has different characteristics from other nonapoptotic forms of cell death, and is mainly characterized by mitochondrial changes including increasing membrane density, mitochondrial atrophy and mitochondrial outer membrane rupture [26]. Ferroptosis critically contributes to the progression of MIRI. It is worth mentioning that ferroptosis is important in not only MIRI, but also other cardiovascular diseases such as myocardial infarction [27], abdominal aortic aneurysm [28] and atherosclerosis [29].
Ischaemia-reperfusion is accompanied by increased production of ROS and reactive nitrogen species [30]. In addition, increase in free iron in cardiomyocytes occurs during the ischaemia-reperfusion [31]. The normal function of cardiomyocytes is based on iron-containing mitochondrial enzymes, and so iron homeostasis is critical for cardiomyocytes [32]. Lipid ROS generation is one of the main features of ferroptosis, and ferroptosis is an iron‐dependent form of RCD. The common factors reflect the complex regulatory relationship among these processes. Currently, some reviews have discussed ferrotopsis as a novel therapeutic target for MIRI, which has received much attention. On the one hand, the induction of ferroptosis can be achieved through different targets, including system Xc−, GPX-4, GSH, ROS, iron and voltage-dependent anion channels. On the other hand, effective ways to inhibit ferroptosis include inhibiting lipid peroxidation, reducing iron concentrations, regulating iron transport, reducing iron intake, increasing GPX4 expression, reducing ROS, removing free radicals and preventing mitochondrial lipid peroxidation [26]. Therefore, finding drugs that exert these effects is a good strategy. In addition, drug studies are urgently needed for the treatment of MIRI.
In the current study, we found that PHC inhibited the expression of ACSL4, and ACSL4 overexpression reversed the effects of PHC on the molecular markers of ferroptosis. It was concluded that PHC inhibited ferroptosis by decreasing the expression of ACSL4 and ultimately suppressing MIRI. This conclusion confirmed our hypothesis.
ACSL4 is closely related to lipid ROS production through PUFAs [33]. Therefore, by inhibiting ACSL4, PHC can reduce the production of lipid ROS and subsequently inhibit ferroptosis. In this study, PHC reduced the levels of ferrous ions cardiomyocytes during MIRI, and promoted the expression of GPX4. These effects play important roles in inhibiting ferroptosis. Although we illustrated the role of PHC in ACSL4-mediated ferroptosis, whether GPX4 is involved in the PHC-mediated inhibition of ferroptosis is still unknown. Furthermore, how PHC affects iron levels in cardiomyocytes is another question. Unfortunately, these questions were not addressed in this study. In several studies, PHC could activate the Nrf2/HO-1 signalling pathway [16, 34], and Nrf2 has been reported to be implicated in the regulation of ferroptosis [25]. HO-1, a downstream target of Nrf2, catalyses the degradation of haem to produce ferrous iron, leading to mitochondrial iron overload, which increases ferroptosis [35, 36]. Therefore, it is possible that PHC alters iron levels through Nrf2/HO-1. In addition, Nrf2 was shown to regulate the expression of GPX4 in a previous study [37]. PHC may also affect GPX through Nrf2. This study showed a decrease in iron and GPX4 levels after PHC treatment. Thus, it is possible that PHC can affect the progression of ferroptosis via Nrf2. However, this is just a matter of speculation and needs to be verified experimentally. In follow-up studies, we will further study PHC to clarify its mechanism of action.
Based on this study and other PHC and MIRI studies, it is clear that PHC inhibits MIRI and can be used as a candidate drug to treat MIRI. This study revealed the inhibitory effect of PHC pretreatment on MIRI, and previous studies revealed that PHC pre/posttreatment alleviated MIRI through different pathways [23, 38]. This proved the consistency of PHC treatment on MIRI inhibition. It is regrettable that the effects of PHC on MIRI or ferroptosis were not validated in a clinical population in our study. Certainly, there is a long way to go before PHC becomes a clinical drug for treating MIRI. However, it is important to highlight some benefits of PHC, which lend support to its potential use as a candidate drug. PHC is a selective anticholinergic drug that mainly acts on muscarinic 1 and muscarinic 3 subtype receptors in the heart, and has little effect on muscarinic 2 subtype receptors. Therefore, PHC does not have the cardiovascular side effect of increasing heart rate [39]. In addition, the half-life of PHC is 10.35 h, and so it has a longer duration of action [40]. Most importantly, PHC has a palliative effect on many diseases and may have the potential to be applied to them.
Our study has several limitations. We were unable to clarify whether GPX4 is involved in PHC-mediated ferroptosis inhibition and the mechanism by which PHC changes iron levels. We also failed to verify the effects of PHC on MIRI and ferroptosis in clinical populations.
In conclusion, this study clarified the role of PHC in ferroptosis, and preliminarily demonstrated that ACSL4 participates in PHC-mediated inhibition of ferroptosis in the cardiomyocytes of MIRI rats. This study is the first to reveal the relationship among PHC, ACSL4, ferroptosis and MIRI. This study also provided a theoretical basis for elucidating the pharmacodynamic mechanism of PHC.
Data Availability
All data used for figures and results are available from the corresponding author on reasonable request.
References
Scirica BM, Bergmark BA, Morrow DA, Antman EM, Bonaca MP, Murphy SA, et al (2020) Nonculprit Lesion Myocardial Infarction Following Percutaneous Coronary Intervention in Patients With Acute Coronary Syndrome. J Am Coll Cardiol 1095–106. https://doi.org/10.1016/j.jacc.2019.12.067.
Sanchez-Hernandez CD, Torres-Alarcon LA, Gonzalez-Cortes A, Peon AN (2020) Ischemia/Reperfusion Injury: Pathophysiology, Current Clinical Management, and Potential Preventive Approaches. Mediators Inflamm 8405370. https://doi.org/10.1155/2020/8405370.
Neri M, Riezzo I, Pascale N, Pomara C, Turillazzi E (2017) Ischemia/Reperfusion Injury following Acute Myocardial Infarction: A Critical Issue for Clinicians and Forensic Pathologists. Mediators Inflamm 7018393. https://doi.org/10.1155/2017/7018393.
Schanze N, Bode C, Duerschmied D (2019) Platelet Contributions to Myocardial Ischemia/Reperfusion Injury. Front Immunol 1260. https://doi.org/10.3389/fimmu.2019.01260.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 1060–72. https://doi.org/10.1016/j.cell.2012.03.042.
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al (2021) Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther 49. https://doi.org/10.1038/s41392-020-00428-9.
Liu J, Kang R, Tang D (2022) Signaling pathways and defense mechanisms of ferroptosis. FEBS J 7038–50. https://doi.org/10.1111/febs.16059.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al (2017) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 91–8. https://doi.org/10.1038/nchembio.2239.
Wang Y, Zhang M, Bi R, Su Y, Quan F, Lin Y, et al (2022) ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol 102262. https://doi.org/10.1016/j.redox.2022.102262.
Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al (2019) Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ 2284–99. https://doi.org/10.1038/s41418-019-0299-4.
Tuo QZ, Liu Y, Xiang Z, Yan HF, Zou T, Shu Y, et al (2022) Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct Target Ther 59. https://doi.org/10.1038/s41392-022-00917-z.
Xu Y, Li X, Cheng Y, Yang M, Wang R (2020) Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J 16262–75. https://doi.org/10.1096/fj.202001758R.
Fan Z, Cai L, Wang S, Wang J, Chen B (2021) Baicalin Prevents Myocardial Ischemia/Reperfusion Injury Through Inhibiting ACSL4 Mediated Ferroptosis. Front Pharmacol 628988. https://doi.org/10.3389/fphar.2021.628988.
Tang LJ, Luo XJ, Tu H, Chen H, Xiong XM, Li NS, et al (2021) Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol 401–10. https://doi.org/10.1007/s00210-020-01932-z.
Xiao HT, Liao Z, Tong RS (2012) Penehyclidine hydrochloride: a potential drug for treating COPD by attenuating Toll-like receptors. Drug Des Devel Ther 317–22. https://doi.org/10.2147/DDDT.S36555.
Liu Z, Li Y, Yu L, Chang Y, Yu J (2020) Penehyclidine hydrochloride inhibits renal ischemia/reperfusion-induced acute lung injury by activating the Nrf2 pathway. Aging (Albany NY) 13400–21. https://doi.org/10.18632/aging.103444.
Wang YP, Li G, Ma LL, Zheng Y, Zhang SD, Zhang HX, et al (2014) Penehyclidine hydrochloride ameliorates renal ischemia-reperfusion injury in rats. J Surg Res 390–7. https://doi.org/10.1016/j.jss.2013.07.041.
Zhang Y, Leng YF, Xue X, Zhang Y, Wang T, Kang YQ (2011) Effects of penehyclidine hydrochloride in small intestinal damage caused by limb ischemia-reperfusion. World J Gastroenterol 254–9. https://doi.org/10.3748/wjg.v17.i2.254.
Yu C, Wang J (2013) Neuroprotective effect of penehyclidine hydrochloride on focal cerebral ischemia-reperfusion injury. Neural Regen Res 622–32. https://doi.org/10.3969/j.issn.1673-5374.2013.07.006.
Zi C, Zhang C, Yang Y, Ma J (2020) Penehyclidine hydrochloride protects against anoxia/reoxygenation injury in cardiomyocytes through ATP-sensitive potassium channels, and the Akt/GSK-3beta and Akt/mTOR signaling pathways. Cell Biol Int 1353–62. https://doi.org/10.1002/cbin.11329.
Yang Y, Zhao L, Ma J (2017) Penehyclidine hydrochloride preconditioning provides cardiac protection in a rat model of myocardial ischemia/reperfusion injury via the mechanism of mitochondrial dynamics mechanism. Eur J Pharmacol 130–9. https://doi.org/10.1016/j.ejphar.2017.07.031.
Lin D, Ma J, Xue Y, Wang Z (2015) Penehyclidine Hydrochloride Preconditioning Provides Cardioprotection in a Rat Model of Myocardial Ischemia/Reperfusion Injury. PLoS One e0138051. https://doi.org/10.1371/journal.pone.0138051.
Lin D, Cui B, Ren J, Ma J (2018) Regulation of VDAC1 contributes to the cardioprotective effects of penehyclidine hydrochloride during myocardial ischemia/reperfusion. Exp Cell Res 257–63. https://doi.org/10.1016/j.yexcr.2018.04.004.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 486–541. https://doi.org/10.1038/s41418-017-0012-4.
Chen X, Li J, Kang R, Klionsky DJ, Tang D (2021) Ferroptosis: machinery and regulation. Autophagy 2054–81. https://doi.org/10.1080/15548627.2020.1810918.
Zhang Y, Xin L, Xiang M, Shang C, Wang Y, Wang Y, et al (2022) The molecular mechanisms of ferroptosis and its role in cardiovascular disease. Biomed Pharmacother 112423. https://doi.org/10.1016/j.biopha.2021.112423.
Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW, et al (2019) Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis 835. https://doi.org/10.1038/s41419-019-2061-8.
Sampilvanjil A, Karasawa T, Yamada N, Komada T, Higashi T, Baatarjav C, et al (2020) Cigarette smoke extract induces ferroptosis in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol H508-H18. https://doi.org/10.1152/ajpheart.00559.2019.
Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al (2019) Atherosclerosis. Nat Rev Dis Primers 56. https://doi.org/10.1038/s41572-019-0106-z.
Lillo-Moya J, Rojas-Sole C, Munoz-Salamanca D, Panieri E, Saso L, Rodrigo R. Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury. Antioxidants (Basel). 2021. https://doi.org/10.3390/antiox10050667.
Nakamura T, Naguro I, Ichijo H (2019) Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim Biophys Acta Gen Subj 1398–409. https://doi.org/10.1016/j.bbagen.2019.06.010.
Paterek A, Mackiewicz U, Maczewski M (2019) Iron and the heart: A paradigm shift from systemic to cardiomyocyte abnormalities. J Cell Physiol 21613–29. https://doi.org/10.1002/jcp.28820.
Zhao WK, Zhou Y, Xu TT, Wu Q (2021) Ferroptosis: Opportunities and Challenges in Myocardial Ischemia-Reperfusion Injury. Oxid Med Cell Longev 9929687. https://doi.org/10.1155/2021/9929687.
Staff PO (2016) Correction: Penehyclidine Hydrochloride Pretreatment Ameliorates Rhabdomyolysis-Induced AKI by Activating the Nrf2/HO-1 Pathway and Allevi-ating Endoplasmic Reticulum Stress in Rats. PLoS One e0154138. https://doi.org/10.1371/journal.pone.0154138.
Kwon MY, Park E, Lee SJ, Chung SW (2015) Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 24393–403. https://doi.org/10.18632/oncotarget.5162.
Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC (2018) Heme oxygenase-1 mediates BAY 11–7085 induced ferroptosis. Cancer Lett 124–37. https://doi.org/10.1016/j.canlet.2017.12.025.
Wang Y, Yan S, Liu X, Deng F, Wang P, Yang L, et al (2022) PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ 1982–95. https://doi.org/10.1038/s41418-022-00990-5.
Tan H, Chen L, Ma J (2017) Penehyclidine hydrochloride post-conditioning reduces ischemia/reperfusion-induced cardiomyocyte apoptosis in rats. Exp Ther Med 4272–8. https://doi.org/10.3892/etm.2017.5089.
Wang YA, Zhou WX, Li JX, Liu YQ, Yue YJ, Zheng JQ, et al (2005) Anticonvulsant effects of phencynonate hydrochloride and other anticholinergic drugs in soman poisoning: neurochemical mechanisms. Life Sci 210–23. https://doi.org/10.1016/j.lfs.2005.04.071.
Wang Y, Gao Y, Ma J (2018) Pleiotropic effects and pharmacological properties of penehyclidine hydrochloride. Drug Des Devel Ther 3289–99. https://doi.org/10.2147/DDDT.S177435.
Acknowledgements
We are grateful for the support from the National Natural Science Foudation of China and the Capital Medical University.
Funding
This study was supported by the National Natural Science Foundation of China (under Grant 82172151).
Author information
Authors and Affiliations
Contributions
Conception and methodology: Duomao Lin and Boqun Cui; Formal analysis: Zeyou Qi and Wenjun Liu; Supervision: Guanzheng Zhang. All authors edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethic Approval
All institutional and national guidelines for the care and use of laboratory animals were followed and approved by the appropriate institutional committees.
No human studies were carried out by the authors for this article.
Conflicts of Interest
The authors have no relevant financial or non-financial interests to disclose.
Disclosures
All authors declare that they have no competing interest.
Additional information
Associate Editor Guoping Li oversaw the review of this article
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lin, D., Cui, B., Qi, Z. et al. A New Aspect of Penehyclidine Hydrochloride in Alleviating Myocardial Ischemia–Reperfusion Injury: Ferroptosis. J. of Cardiovasc. Trans. Res. 16, 1373–1382 (2023). https://doi.org/10.1007/s12265-023-10420-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-023-10420-7