
Abstract
Cardiac hypertrophy is an adaptive response to abnormal physiological and pathological stimuli, which can be classified into concentric and eccentric hypertrophy, induced by pressure overload or volume overload, respectively. In both physiological and pathological scenarios, females generally show a more favorable form of hypertrophy compared with their male counterparts. However once established, cardiac hypertrophy is a stronger risk factor for heart failure in females. Pre-menopausal women are better protected against cardiac hypertrophy compared with men, but this protection is abolished following menopause and is partially restored after estrogen replacement therapy. Estrogen exerts its protection by counteracting pro-hypertrophy signaling pathways, whereas androgen mostly plays an opposite role in cardiac hypertrophy. We here summarize the progress in the understanding of sexual dimorphisms in cardiac hypertrophy and highlight recent breakthroughs in the regulatory role of sex hormones and their intricate molecular networks, in order to shed light on gender-oriented therapeutic efficacy for pathological hypertrophy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease is the leading global cause of death [1]. Nearly all types of cardiovascular disease are associated with cardiac hypertrophy [2]. Cardiac hypertrophy is an increase in heart mass or an enlargement of the heart muscle [3], which can be broadly divided into two types: physiological hypertrophy and pathological hypertrophy. Physiological hypertrophy generally develops in regular physical activity including growth and pregnancy, or chronic exercise training such as aerobic exercise and strength training, and maintains normal heart function [4]. In contrast, pathological cardiac hypertrophy is caused by various adverse stimuli such as hypertension, valvular stenosis or regurgitation, and obesity [4, 5]. Either physiological or pathological hypertrophy is an adaptive response to mechanical stimuli [3]. Mechanical stimuli can be classified as pressure overload and volume overload [6]. Pressure overload induces concentric cardiac hypertrophy, whereas volume overload produces eccentric cardiac hypertrophy. As for physiological hypertrophy, aerobic exercise and pregnancy result in volume overload and eccentric hypertrophy, while strength training leads to a pressure load and concentric hypertrophy. In pathological hypertrophy, hypertension and aortic stenosis produce concentric hypertrophy due to an increase in systolic wall stress, while valvular regurgitation and arteriovenous fistula produce eccentric hypertrophy due to an increase in diastolic wall stress [7]. Notably, there is overlap between the mechanisms of physiological and pathological cardiac hypertrophy, for it is evidenced that some signaling effectors play an essential role in both physiological and pathological hypertrophy [3, 8, 9].
In recent years, a growing number of preclinical and clinical studies have clearly indicated the important role of gender in cardiac hypertrophy. Previous clinical studies have shown that cardiac hypertrophy appears later in women than it does in men; however once established, cardiac hypertrophy is a stronger risk factor for heart failure (HF) in women than in men [10]. To interrogate the mechanism underlying the discrepancy, some studies believe that it is mainly attributed to sex hormones such as estrogen and androgen and their receptors [11, 12]. The ground for the involvement of sex hormones is that the incidence of cardiomyopathies is lower in premenopausal women than in age-matched men, whereas this trend disappears when it comes to postmenopausal women [13, 14]. Here, we review the literature comparing the gender differences in pathological and physiological cardiac hypertrophy in humans as well as in animal models, and further discuss the underlying functional characteristics of sex hormones and their intricate molecular networks.
Gender Differences in Pathological Cardiac Hypertrophy—Human Studies
The most frequent precursor and facilitator of heart failure is pathological cardiac hypertrophy, due to hypertension or other non-ischemic origins in women, whereas due to an ischemic etiology in men [10]. In patients with severe aortic stenosis, the ventricular remodeling is more severe in men, with more inflammatory response and fibrosis, resulting in a poorer prognosis than that of women [15]. Another clinical study, the CURRENT AS study, has revealed that a longer 5-year survival rate is shown in women rather than men with severe aortic valve stenosis [16]. Also, in aortic stenosis, after aortic valve replacement, the left ventricular (LV) ejection fraction in women is significantly higher than men, indicating a better adaptation to long-term mechanical stress loads in women [17]. In mitral regurgitation induced by valve prolapse, women show benign presentation by more often with thick leaflets and less often with flail, posterior leaflet prolapse or severe regurgitation [18]. Interestingly, in obesity-induced cardiac hypertrophy, compared with obese females who showed a combination of concentric and eccentric cardiac hypertrophy, obese males predominantly present concentric hypertrophy [19]. Noticeably, it is previously reported that cardiovascular mortality of concentric cardiac hypertrophy was higher than that of eccentric hypertrophy [6, 20]. Given that concentric hypertrophy is more strongly related to cardiovascular mortality than eccentric hypertrophy [19], those observations may explain the observed gender difference that obese women have lower cardiovascular risk than obese men.
Although women usually show higher adaptability in pathological cardiac hypertrophy, cardiac hypertrophy itself would impair the gender advantage in women. Recent studies have pointed out that in women with severe hypertension-induced LV hypertrophy, the mechanical overload caused by blood pressure will offset the gender-related benefits in cardiovascular risk [21]. Compared with men, women with cardiovascular disease hold a greater proportion of diastolic dysfunction if they have progressed into heart failure with preserved ejection fraction (HFpEF) [22], which suggests that the protective effect in the development of cardiac hypertrophy shown in female patients is not obvious in the end stage of pathological cardiac hypertrophy or the heart failure stage.
Gender Differences in Pathological Cardiac Hypertrophy—Experimental Studies
Gender factors have been systematically investigated in most animal models, including rodents, rabbits, and dogs, but the mechanisms responsible for the sex differences in hypertrophy have been studied mainly in rodents. The mechanical load in animal models usually includes pressure overload or volume overload, which simulates the process of human cardiac hypertrophy under persistent pathological hemodynamic stimulation.
Pressure Overload Model
Chronic pressure overload produced by transverse aortic constriction (TAC) triggers the progression from compensatory hypertrophy to heart failure [23]. It has been indicated that male but not female rats show an early heart failure phenotype characterized by ventricular dilatation after 20-week TAC [24]. Other studies have also reported that male mice show a higher proportion of pathological cardiac hypertrophy, fibrosis, apoptosis, and heart failure than female mice in chronic stress loads, and female mice possess more potent cardiac reserve under similar LV hypertrophy and systolic wall tension [25]. Further investigations using male and female rats with banded abdominal aorta to induce pressure overload have revealed that there is no significant sex difference in the early stage (6 weeks) of LV hypertrophy after banding. However, after 12 weeks, male rats show a decrease in myocardial contractility, accompanied by increased myocardial wall stiffness and impaired diastolic function, while female rats show no deterioration of LV structure and function [26].
During pressure overload, cardiomyocyte apoptosis is more augmented in male mice, which indicates that the molecular level of gender differences in cardiac hypertrophy is consistent with the morphological animal level [25]. In addition, signaling pathways involved in cardiac hypertrophy are also differentially expressed between males and females. The expression of Ca2+/calmodulin-dependent protein kinase phosphatase (CaMKP), a key protein in the development of cardiac hypertrophy, increases in male rats while decreased in female mice after TAC [27].
Volume Overload Model
Unlike pressure overload, volume overload induces eccentric hypertrophy due to long-term stimulation of the diastolic ventricular wall by a large amount of blood flow into the ventricular cavity. The animal models used for volume overload study are created by arteriovenous shunt (AV), or novelly, by aortic insufficiency (AI) [6, 28]. The gender-related specific characteristics found in volume overload model of AV are also consistent with those reported in pressure overload. After 8 weeks of AV, the mortality of male rats was more than 10 times higher than that of female counterparts, and male rats showed more significant ventricular dilatation and lower ventricular compliance [29]. Hemodynamic analysis also unveiled that an increase in left ventricular end-diastolic pressure (LVEDP) as well as a decrease in fractional shortening occurred in males only after 4 weeks of AV, and the difference was more pronounced with time after surgery [30]. However, in the AI model, despite similar regurgitation severity, female rats developed more LV eccentric hypertrophy in response to chronic AI than males [31, 32]. AI seems to impose a greater LV workload on females relative to their smaller body and heart size [31]. Intriguingly, the gene expression of fatty acid oxidation (FAO)-related LV enzyme activity and mitochondrial function decreased only in male AI rats, while remained closer to normal in female ones; therefore, female rats still kept higher cardiac reserve to tolerate persistent severe AI [32].
An increase in apoptosis in the male heart and a reduction in females were observed at 16 weeks of AV operation, characterized by prominent increases in the pro-apoptotic proteins such as BAX, caspases 3 and 9 in males [33]. In addition, the β-receptor expression in the volume overloaded heart was also different between genders. Though the β1-receptor affinity had no difference in male and female rats, the AV-induced increase in β1-receptor density in female rats was higher than that in males, and the levels of adrenaline and norepinephrine in plasma were lower in females [34]. Thus, in the activated cardiac β-receptor system due to volume overload, excessive catecholamine leads to lower β-receptor level in male rats, which makes a poor compensation to cardiac remodeling compared with female rats [34].
Role of Sex Hormones in Pathological Cardiac Hypertrophy
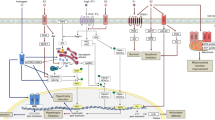
Based on the strong link between gender and pathological cardiac hypertrophy, it is speculated that the gender differences in pathological cardiac hypertrophy could be attributed to sex hormones and their receptors [35, 36]. Indeed, estrogen and androgen have been found to exert different effects on the heart and peripheral circulation in human and rodents (Table 1) [45, 49]. In addition, several clinical trials indicate that hormone replacement therapy reduces cardiovascular risk if it starts within the first years after menopause [50,51,52]. Thus, sex hormones are critically involved in cardiac hypertrophy-related signaling pathways (Fig. 1) [13, 51].

Estrogen and androgen modulated signaling pathways in pathological cardiac hypertrophy. Estrogen mediates its anti-hypertrophic effects by binding to its receptors, ERα and ERβ. ER stimulation under pathological stimuli attenuates MEK1/2-ERK1/2-Elk1 signaling cascade, to ameliorate hypertrophic genes reprogramming and hypertrophic response. ER induces high MKP-1 levels to reduce p38 activation and attenuate hypertrophy. ER abrogates calcineurin-mediated hypetrophic effects through a NFAT-MEF2/GATA4 mechanism. ER activates PI3K/Akt pathway, as well as PI3K/MCIP1 signaling leading to compromised calcineurin expression. ER induces eNOS-sGC-cGMP-PKGIα signaling cascade providing cardioprotection against cardiac hypertrophy. Compared with estrogen/ER, androgen/AR shows a mostly opposite effect in pathological cardiac hypertrophy. Besides MEK1/2-ERK1/2-Elk1 and eNOS-sGC-cGMP-PKGIα signaling cascades, AR promotes calcium handling protein activity through IP3 and CaMKII stimulation and GSK-3β inhibition. AR, androgen receptor; Akt, protein kinase B; CaMKII, Ca2+/calmodulin-dependent protein kinase II; cGMP, cyclic guanosine monophosphate; eNOS, endothelial nitric oxide synthase; ERα/β, estrogen receptor α or β; ERK1/2, extracellular signal-regulated kinase 1/2; Elk1, E26 transformation-specific like-1; GSK3β, glycogen synthase kinase 3 β; GATA4, GATA binding protein 4; IP3/Ca2+, inositol 1,4,5-trisphosphateinositol-mediated Ca2+; MEK1/2, MAP kinase/ ERK kinase 1/2; MMP2, matrix metalloprotease 2; MCIP, myocyte-enriched calcineurin interactin protein; MKP-1: MAPK phosphatase-1; MEF2, myocyte enhancer factor 2; mTORC1, mammalian target of rapamycin complex 1; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor of kappa light chain gene enhancer in B cells; PGC-1α, peroxisome proliferative activated receptor gamma coactivator 1 alpha; PI3K, phosphatidylinositol 3-kinase; PKGIα, protein kinase Iα; S6K1, 40S ribosomal protein S6 kinase 1; sGC, soluble guanylyl cyclase; hypertrophic gene program, such as ANP, BNP, CTGF
Estrogen and Its Cardioprotective Role
Population-based studies have found that postmenopausal women show higher risk of cardiovascular disease than non-menopausal women under the same age and women with early menopause have higher cardiovascular risk than those with late menopause [50]. Moreover, women who initiate hormone (conjugated equine estrogen) replacement therapy closer to menopause tend to have reduced risk of cardiovascular disease, compared with the increased risk among women more distant from menopause [51]. Another clinical trial also using conjugated equine estrogen for hormone replacement therapy demonstrates that it contributes to the reduction of LV mass in hypertensive postmenopausal women, although the effect does not appear to be associated with changes in growth-promoting factors, such as blood pressure, serum angiotensin-converting enzyme activity, plasma aldosterone, and insulin resistance [52].
Consistent with the clinical studies that have demonstrated the postmenopausal loss of cardioprotection in women, many preclinical studies have shown that the ability to maintain better cardiac function is offset by bilateral ovariectomy in female rats under pressure overload, recapitulating that estrogen is vital in delaying the progress of cardiac hypertrophy under pressure overload [37]. Moreover, in pressure overload mice, 17beta-estradiol (E2) delays the occurrence of cardiac hypertrophy and prevents fibrosis in the heart by an estrogen receptor-dependent mechanism [35, 36]. A recent study even reported that E2 rescued pre-existing HF by stimulating angiogenesis and suppressing fibrosis [38], although van Eickels et al. reported that E2 attenuated the hypertrophic response but not cardiac fibrosis and cell proliferation in the heart [37]. In volume load models, estrogen and phytoestrogen effectively resist adverse left ventricular remodeling and play a cardioprotective role in female rats after AV [29, 42]. Even in male rats, supplement of estrogen attenuates left ventricular dilatation and maintains function in AV rats [39].
Mitogen-Activated Protein Kinase (MAPK) Pathway
It is documented that 17beta-estradiol (E2) inhibits AngII-induced cardiac hypertrophy and fibrosis in female mice by inactivation of extracellular signal-regulated kinase (ERK), as well as by stimulation of brain natriuretic peptide to attenuate interstitial fibrosis [40]. E2 replacement also reduces p38 activation and attenuates hypertrophy induced by pressure overload in ovariectomized mice [37]. Mechanistically, in norepinephrine-induced hypertrophy, E2 induces high MAPK phosphatase-1 (MKP-1) levels, which precluded p38 activation, suggesting that p38 activation is critically involved in norepinephrine-induced hypertrophy [53]. A recent study also found that E2 was able to protect cardiomyocytes from AngII-induced injury through upregulation of Sirtuin 1 and activation of adenosine monophosphate-activated protein kinase (AMPK) [54].
eNOS Pathway
A study using ovariectomized female rats showed that ovariectomy augmented pressure overload-induced cardiac hypertrophy, deteriorated heart function, and increased mortality, which could be attributed to impairment of endothelial nitric oxide synthase (eNOS) and Akt activities by ovariectomy [55]. Notably, PDE5 is overexpressed in cardiac hypertrophy, and PDE5 inhibitors such as sildenafil have provided cardioprotection against cardiac hypertrophy in experimental and clinical studies, via a mechanism involving estrogen, in an eNOS-soluble guanylyl cyclase (sGC)-cyclic guanosine monophosphate (cGMP)-PKGIα (protein kinase Iα) pathway [56]. This mechanism may explain why a large clinical trial testing the efficacy of sildenafil in patients with heart failure failed [57], for it mainly enrolled male patients, whereas sildenafil ameliorates pressure overload cardiac hypertrophy through the aforementioned estrogen-dependent mechanism in females but not males.
Extracellular Signaling
The efficacy of estrogen treatment in chronic volume overload was associated with a reduction in oxidative stress and circulating endothelin-1 levels, as well as prevention of matrix metalloproteinase-2 and -9 activation and breakdown of collagen in the early stage of remodeling [39]. Of note, a study further showed that low levels of estrogen produced cardiomyocyte hypertrophy through ERK1/2/sodium-hydrogen exchange isoform 1 (NHE-1) activation and intracellular alkalinization whereas an antihypertrophic effect was seen at high concentrations [41].
Calcium-Related Signaling
In consistence with the different hypertrophic response to pressure or volume overload between females and males, a sexually dimorphic role for estrogen in the modulation of signaling activity in cardiac myocyte has been well documented. Cardiac hypertrophy is characterized by abnormality of intracellular Ca2+ homeostasis, as well as Ca2+ activation of two major hypertrophic signaling pathways, the Ca2+/calmodulin-dependent protein kinase II (CaMKII)-histone deacetylase (HDAC)-myocyte enhancer factor 2 (MEF2) pathway and the calcineurin (CaN)-nuclear factor activation transcription (NFAT) pathway, both in turn enhances hypertrophy or apoptosis of cardiomyocytes [36, 58]. Thus, several studies have investigated the regulation of calcium-related signaling by estrogen [35, 36]. Estrogen receptor (ER) knockout (ERKO) mice show increased expression of L-type calcium channel-related molecules at mRNA and protein levels in ventricular myocytes [59], while estrogen also reduces the activity of L-type calcium channels in vitro [60]. TAC was associated with increased CaMKP expression in male LV whereas it tended to be decreased in females, leading to differences in CaMKII and MEF2 activation between sexes [27]. On the other hand, two independent groups reported that estrogen reduced pressure overload-induced hypertrophy by an ER-dependent mechanism that increased calcineurin degradation, depressed expression of NFAT [35, 36]. Moreover, modulatory calcineurin-interacting protein (MCIP1), an anti-hypertrophy protein which directly inhibits calcineurin expression, is induced by estrogen [40]. Therefore, estrogen plays an important role in inhibiting the development of cardiac hypertrophy through regulation of Ca2+-related pathways. Even so, considering published data concerning regulation of Ca2+ handling proteins by estrogen in volume overload is still in paucity, more relevant studies are warranted to elucidate their reciprocal effects.
Estrogen Receptor
An ERKO model has been further used to elucidate the protective mechanisms by which estrogen exerted in pressure overload. There are two subtypes of estrogen receptors, ERα and ERβ. TAC was performed after knocking out the corresponding subtypes of female mice. Compared with WT female mice, the heart weight to body weight ratio in ERα knockout female mice was similar, but in ERβ knockout female mice, the ratio was significantly increased, implying that the cardiac protection of estrogen under pressure overload stimulation is mediated by ERβ [25, 61, 62].
Androgen and Its Pro-hypertrophic Role
Compared with the mostly cardioprotective role of estrogen in pathological cardiac hypertrophy, androgen, mainly characterized by testosterone and its highly active metabolite dihydrotestosterone (DHT), shows an opposite effect in cardiac hypertrophy [63]. By binding to androgen receptor (AR), androgen exerts a pro-hypertrophic effect on cardiomyocytes [63]. In preclinical studies, testosterone is considered to play a critical role to mediate cardiac hypertrophy and fibrosis induced by AngII in male rats [48]. Anti-androgenic therapy with finasteride (an antiandrogen) or relevant androgen receptor antagonists has exhibited attenuation on cardiac hypertrophy, fibrosis, and dysfunction in not only male, but also in female mice [64, 65]. In women, low total testosterone concentration is associated with a decreased risk of prevalent hypertension [66]. The above findings indicate that androgen-AR system participates in normal cardiac growth and modulates cardiac adaptive hypertrophy under hypertrophic stress. However, the role of androgen in males seems much more complicated than in females, and testosterone as a supplement to treat heart failure is controversial, for it produces a few successes in some clinical trials but not in others [67,68,69]. In men, testosterone level begins to decrease after age 40, and this decrease is associated with an increase in all-cause mortality and cardiovascular risk [70]. In male mice, testosterone deficiency leads to intracellular Ca2+ dysregulation and myofilament dysfunction, which facilitates diastolic dysfunction in the setting of aging [71]. Intriguingly, when AR is systematically deleted, male mice show impairments of both the concentric hypertrophic response and LV function and an exacerbation of cardiac fibrosis under AngII stimulation [72]. Recently, the biphasic effects of testosterone have been found in male rats during a time-specific testosterone administration, in which physiological cardiac hypertrophy is apparent with an upregulation of α-MHC, and without any change in myofilament contractile activation after 4-week treatment of testosterone, whereas pathological cardiac hypertrophy was observed as indicated by suppression of myofilament activation and augmentation of myocardial collagen deposition without transition of MHC isoforms after 8- and 12-week testosterone treatment [45]. The biphasic effects of testosterone may partially explain why testosterone replacement treatment shows contradictory results in clinical trials.
Molecular Mechanism of Androgen Regulation
It is recently found that Ca2+ handling proteins are crucially involved in testosterone-induced cardiac hypertrophy. Cardiac myocyte hypertrophy induced by testosterone involves a cooperative mechanism that links androgen signaling with the recruitment of NFAT through activation of calcineurin and inhibition of glycogen synthase kinase-3beta (GSK-3β), a negative regulator of calcineurin [46]. Moreover, androgen induces cardiomyocyte hypertrophy through activation of MEF2 mediated by CaMKII [47]. The mammalian target of rapamycin complex 1 (mTORC1) is a major regulator of cell growth. It is documented that testosterone activates the mTORC1/40S ribosomal protein S6 kinase 1 (S6K1) axis through 1,4,5-trisphosphate (IP3)/Ca2+ and MEK/ERK1/2 to induce cardiomyocyte hypertrophy [44]. Androgens also regulate the eNOS-sGC-cGMP-PKGIα pathway, for cardiac hypertrophy, and fibrosis is significantly more pronounced in males compared with females of GC-A knockout mice, and the gender differences are virtually abolished by castration or targeted deletion of the AngII type 1A receptor gene (AT1A), suggesting that androgen contributes to gender-related differences in cardiac hypertrophy and fibrosis by a mechanism involving GC-A and AT1A receptors [43, 73].
Gender Differences in Physiological Cardiac Hypertrophy—Human Studies
In remarkable contrast with maladaptive pathological cardiac hypertrophy, physiological cardiac hypertrophy is adaptive, reversible, and occurs during maturation (normal postnatal growth), exercise, and pregnancy, without morbid effect on cardiac function [7]. Although cardiac output is enhanced to meet the increased metabolic demands, physiological hypertrophy remains adaptive and does not involve cardiac dysfunction and fibrosis [49, 74].
There is no difference in the heart between men and women before puberty, which suggests that males and females are born with the same number and same size of cardiac myocytes [75]. After puberty, women maintain the same heart mass and number of heart muscle cells, but men present a 15–30% increase in heart weight, suggesting a higher hypertrophic potential in men [76]. However, LV diameter increases with aging and results in a loss of myocardial mass only in men, but not in women. The loss of myocardium in older men is attributed to a reduced number of myocytes [76]. Correspondingly, systolic function decreases with aging only in men, whereas decreased diastolic function is found both in men and women [77].
Gender differences in physiological cardiac hypertrophy have not been examined widely, partly due to most of the human studies on the relation between exercise and cardiac hypertrophy are limited to male athletes. Existing studies have shown contradictory results with different sex ratios, recruitment standards, and intervention methodologies. Sullivan et al. found that there was no intergroup difference in stroke volume index, LV ejection fraction, and LV end-diastolic or end-systolic volume indexes between men and women, after upright exercise [78]. Petersen and colleagues reported that young adult elite athletes of both sexes exhibited a significant but similar hypertrophic response to exercise training (by LV and RV volume and mass indices), in accordance with the benign nature of the hypertrophy associated with athlete’s heart [79]. Nevertheless, Higginbotham and colleagues found that LV ejection fraction increased more in men than women, and end-diastolic volume increased by 30% in women but was unchanged in men, with a resultantly similar stroke volume and cardiac output for men and women [80]. Although most clinical trials report that cardiac adaptation to exercise are not statistically different between males and females, considering training intensity influences exercise-induced cardiac remodeling, the same range of LV mass with lower training intensity would implicate higher rate of hypertrophic response in women [81].
Physiological cardiac hypertrophy induced by exercise training is characterized by enhanced fatty acid and glucose oxidation among young healthy physically trained male volunteers [82], and the main metabolic attribute of physiologic hypertrophy is linked with increased cardiac free fatty acid (FFA) uptake and utilization [83]. Consistently, lipolytic activity to catecholamine infusion in women is higher compared to men [84]. Moreover, females oxidize proportionately more lipid and less carbohydrate during exercise compared with males both pre- and post-training [85]. The more enhanced FFA cardiac uptake, which is exercise dependent and catecholamine-induced, implies that FFAs are the predominant cardiac fuel during exercise in females, and also points toward the sex-specific differences in the development of exercise-induced hypertrophy in humans [86].
Gender Differences in Physiological Cardiac Hypertrophy—Experimental Studies
Animal models, especially rodents, provide an excellent tool to study sex-specific differences in the development of physiological cardiac hypertrophy [81]. Treadmill running, freewheel running, and swimming are commonly used to induce physiological hypertrophy [87]. Most of animal studies published so far implicate a more pronounced exercise-induced hypertrophic response shown in females than males. It is found that female mice run much longer and faster than males in voluntary cage running systems, independent of strain and age [7, 9]. Also, female mice develop greater cardiac hypertrophy than their male counterparts [9]. Coherently, treadmill running also causes similar sex-related effects in mice, in which females demonstrate more robust cardiac growth than male counterparts [88]. Moreover, female rats undergoing swim training have more physiological hypertrophy than male rats [89, 90].
In agreement with human studies, cardiac expression profiling studies also reveal increased expression of genes involved in FFA metabolism in exercise-induced hypertrophy [91]. It is found that the augmented cardiac hypertrophic responses after exercise training in female rodents are associated with a more increase in plasma FFA levels and a counterbalancing decrease of cardiac glucose uptake [88].
Role of Sex Hormones in Physiological Cardiac Hypertrophy
The anatomical arrangement of the heart is identical between males and females, whereas the size of the female heart is smaller than the age-matched male heart, and the hypertrophic responses are more augmented in females, as discussed above. Therefore, the cardiac response under physiological stimuli is likely influenced by sex-specific hormonal factors. Potential mechanisms underlying this sexual dimorphism in physiological cardiac hypertrophy have been recently proposed, recapitulating the involvement of sex hormones as direct modifiers of hypertrophic response (Fig. 2).

Estrogen and androgen modulated signaling pathways in physiological cardiac hypertrophy. In the context of physiological stimuli, ER stimulation induces ERK1/2-S6 signaling cascade to promote reprogramming of physiologically hypertrophic genes, leading to increased protein synthesis and cardiomyocyte growth. Likewise, ER was necessary for the activation of PI3K/Akt and p38 pathways, leading to higher transcriptional activation of hypertrophy-associated genes. Moreover, estrogen contributes to the sexual dimorphism in physiological hypertrophy through PPARs and PGC-1 pathways to maintain the dynamic capacity of the fatty acid oxidation to increase fatty acid utilization. Androgen induces a physiological cardiac hypertrophy while the underlying mechanism remains to be elucidated. In addition, E2 can be physically synthesized from testosterone by aromatases. Akt, protein kinase B; ERα/β, estrogen receptor α or β; ERK1/2, extracellular signal-regulated kinase 1/2; p38-MAPK, p38-mitogen-activated protein kinase; PPARs, peroxisome proliferator-activated receptors; PGC-1α, peroxisome proliferative activated receptor gamma coactivator 1 alpha; PI3K, phosphatidylinositol 3-kinase; S6, 40S ribosomal protein S6; hypertrophic gene program, such as α-MHC, MEF2, NRF-1/NRF-2
Estrogen and ER
The role of global deficiency of estrogen has been investigated in aromatase knockout (ArKO) mice. ArKO mice lack an enzyme essential for production of estrogen, thus circulating and locally produced estrogen is absent [92]. Haines et al. first investigated cardiac hypertrophic response in ArKO mice [93]. They found that although freewheel running performance was blunted in ArKO females, exercise-induced physiological hypertrophy was unaffected by the global absence of estrogens in either sex [93]. In clinical practice, aromatase inhibitors did not show a significant increase in cardiovascular events [94]. Thus, it seems that although global absence of estrogen clearly influences running behaviors, it does not influence cardiac growth in response to physiologic stimuli.
On the contrary, Dworatzek and colleagues presented a more specific and in-depth study to elucidate the roles of the subtypes of ER in physiological cardiac hypertrophy [9]. They found that the increase in LV mass related to tibia length (LVM/TL) was more pronounced in female mice than in male mice after freewheel running. Coherently, exercise-induced cardiomyocyte area increased only in female mice. More specifically, they found that ERα is involved in running behavior in female mice and ERβ is involved in physiological hypertrophy in both sexes. They in female rat hearts further demonstrated that ERβ was necessary for the activation of PI3K/Akt, ERK1/2/40S ribosomal protein S6 (S6), p38 pathways, as well as an increased expression of key regulators of mitochondrial function and mitochondrial respiratory chain proteins (complexes I, III, and V) after treadmill running. Mechanically, ERβ modulates higher mitochondria dynamics, which leads to increased mitochondrial mass to cope with increased cardiomyocyte size and a more efficient metabolism in mitochondria in the exercise-induced hypertrophy only in female mice [9]. For cardiac expression of ERα is similar in both genders, whereas ERβ expression is significantly higher in females [95], ERβ may play a more significant role in sex differences in physiological hypertrophy. Nevertheless, it is worthwhile to note that ERs are not only activated by estrogens but also by other ligands like growth factors [96]. Thus, the effects of ERs cannot be easily considered as the same as that of estrogen.
Estrogen is also mechanistically indicated to have a positive impact on energy metabolism. Estrogen upregulates lipid utilization and down-regulates glucose oxidation [7]. Besides, estrogen interacts with peroxisome proliferator-activated receptors (PPARs) and PPAR gamma coactivator 1 (PGC-1) [7]. PPARα and its heterodimeric partner, retinoid X receptor (RXR), are key to maintain the dynamic capacity of the FAO to increase fatty acid utilization [97]. Specifically, PGC-1 is in a central position in the most relevant female network of metabolism [98]. In summary, it is suggested that estrogen contributes to the sexual dimorphism in physiological hypertrophy by the modulation of metabolism through PPARs and PGC-1 pathways [97].
Androgen and AR
Research on the role androgen in gender differences in physiological cardiac hypertrophy is sparse. One piece of implication comes from a study using gonadectomized male rats, in which swimming does not induce hypertrophic response in those animals, but testosterone replacement restored ventricular V1 myosin levels to or above normal. Correspondingly, one important concern of testosterone action in patients is induction of cardiac hypertrophy. Testosterone and androgenic anabolic steroids have been misused for enhancement of physical performance despite that they could cause cardiac sudden death [45]. It is recently found that short-term (4 weeks) use of testosterone induces a physiological cardiac hypertrophy by an upregulation of α-MHC without any change in myofilament contractile activation in rats [45]. Intriguingly, E2 can be synthesized from testosterone by aromatases in the heart locally, indicating the synergistic and complicated effects of androgen and estrogen in physiological hypertrophy [99].
Conclusions
Human and animal studies have indicated that cardiac sexual dimorphisms exist in pathological and physiological hypertrophy from the level of cardiac myocyte to the whole heart. Females seem to be prone to concentric hypertrophy, characterized by better cardiac function, compared with their male counterparts. The sexual dimorphisms are partly due to the mostly opposite effects of sexual hormones such as estrogen and androgen, although contradictory results from hormone replacement therapy exist. Since pathological cardiac hypertrophy is an independent risk factor for heart failure in patients, whereas physiological hypertrophy is cardioprotective or indispensable for normal function, a better understanding of the mechanisms through which gender differentially regulates cardiac hypertrophy could open the way to innovative and personalized therapeutic strategies for heart failure.
References
Roth, G. A., Johnson, C., Abajobir, A., Abd-Allah, F., Abera, S. F., Abyu, G., et al. (2017). Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. Journal of the American College of Cardiology, 70(1), 1–25. https://doi.org/10.1016/j.jacc.2017.04.052.
Zhou, L., Ma, B., & Han, X. (2016). The role of autophagy in angiotensin II-induced pathological cardiac hypertrophy. Journal of Molecular Endocrinology, 57(4), R143–R152. https://doi.org/10.1530/JME-16-0086.
Nakamura, M., & Sadoshima, J. (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nature Reviews. Cardiology, 15(7), 387–407. https://doi.org/10.1038/s41569-018-0007-y.
McMullen, J. R., & Jennings, G. L. (2007). Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure. [Research Support, Non-U.S. Gov't Review]. Clinical and Experimental Pharmacology & Physiology, 34(4), 255–262. https://doi.org/10.1111/j.1440-1681.2007.04585.x.
Wu, J., You, J., Wang, S., Zhang, L., Gong, H., & Zou, Y. (2014). Insights into the activation and inhibition of angiotensin II type 1 receptor in the mechanically loaded heart. [Research Support, Non-U.S. Gov't]. Circulation Journal, 78(6), 1283–1289.
You, J., Wu, J., Zhang, Q., Ye, Y., Wang, S., Huang, J., et al. (2018). Differential cardiac hypertrophy and signaling pathways in pressure versus volume overload. American Journal of Physiology. Heart and Circulatory Physiology, 314(3), H552–H562. https://doi.org/10.1152/ajpheart.00212.2017.
Bernardo, B. C., Weeks, K. L., Pretorius, L., & McMullen, J. R. (2010). Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. [Research Support, Non-U.S. Gov't Review]. Pharmacology & Therapeutics, 128(1), 191–227. https://doi.org/10.1016/j.pharmthera.2010.04.005.
Wende, A. R., O'Neill, B. T., Bugger, H., Riehle, C., Tuinei, J., Buchanan, J., et al. (2015). Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes. Molecular and Cellular Biology, 35(5), 831–846. https://doi.org/10.1128/MCB.01109-14.
Dworatzek, E., Mahmoodzadeh, S., Schubert, C., Westphal, C., Leber, J., Kusch, A., et al. (2014). Sex differences in exercise-induced physiological myocardial hypertrophy are modulated by oestrogen receptor beta. Cardiovascular Research, 102(3), 418–428. https://doi.org/10.1093/cvr/cvu065.
Regitz-Zagrosek, V., & Seeland, U. (2011). Sex and gender differences in myocardial hypertrophy and heart failure. Wiener Medizinische Wochenschrift (1946), 161(5–6), 109–116. https://doi.org/10.1007/s10354-011-0892-8.
Blenck, C. L., Harvey, P. A., Reckelhoff, J. F., & Leinwand, L. A. (2016). The Importance of Biological Sex and Estrogen in Rodent Models of Cardiovascular Health and Disease. Circulation Research, 118(8), 1294–1312. https://doi.org/10.1161/CIRCRESAHA.116.307509.
Regitz-Zagrosek, V., & Kararigas, G. (2017). Mechanistic Pathways of Sex Differences in Cardiovascular Disease. Physiological Reviews, 97(1), 1–37. https://doi.org/10.1152/physrev.00021.2015.
Patrizio, M., & Marano, G. (2016). Gender differences in cardiac hypertrophic remodeling. Annali dell'Istituto Superiore di Sanità, 52(2), 223–229. https://doi.org/10.4415/ANN_16_02_14.
Song, J. J., Ma, Z., Wang, J., Chen, L. X., & Zhong, J. C. (2019). Gender Differences in Hypertension. Journal of Cardiovascular Translational Research. https://doi.org/10.1007/s12265-019-09888-z.
Kararigas, G., Dworatzek, E., Petrov, G., Summer, H., Schulze, T. M., Baczko, I., et al. (2014). Sex-dependent regulation of fibrosis and inflammation in human left ventricular remodelling under pressure overload. European Journal of Heart Failure, 16(11), 1160–1167. https://doi.org/10.1002/ejhf.171.
Toyofuku, M., Taniguchi, T., Morimoto, T., Yamaji, K., Furukawa, Y., Takahashi, K., et al. (2017). Sex Differences in Severe Aortic Stenosis- Clinical Presentation and Mortality. Circulation Journal, 81(8), 1213–1221. https://doi.org/10.1253/circj.CJ-16-1244.
Rohde, L. E., Zhi, G., Aranki, S. F., Beckel, N. E., Lee, R. T., & Reimold, S. C. (1997). Gender-associated differences in left ventricular geometry in patients with aortic valve disease and effect of distinct overload subsets. The American Journal of Cardiology, 80(4), 475–480.
Avierinos, J. F., Inamo, J., Grigioni, F., Gersh, B., Shub, C., & Enriquez-Sarano, M. (2008). Sex differences in morphology and outcomes of mitral valve prolapse. Annals of Internal Medicine, 149(11), 787–795.
Rider, O. J., Lewandowski, A., Nethononda, R., Petersen, S. E., Francis, J. M., Pitcher, A., et al. (2013). Gender-specific differences in left ventricular remodelling in obesity: insights from cardiovascular magnetic resonance imaging. European Heart Journal, 34(4), 292–299. https://doi.org/10.1093/eurheartj/ehs341.
Krumholz, H. M., Larson, M., & Levy, D. (1995). Prognosis of left ventricular geometric patterns in the Framingham Heart Study. Journal of the American College of Cardiology, 25(4), 879–884. https://doi.org/10.1016/0735-1097(94)00473-4.
Gerdts, E., Izzo, R., Mancusi, C., Losi, M. A., Manzi, M. V., Canciello, G., et al. (2018). Left ventricular hypertrophy offsets the sex difference in cardiovascular risk (the Campania Salute Network). International Journal of Cardiology, 258, 257–261. https://doi.org/10.1016/j.ijcard.2017.12.086.
Gori, M., Lam, C. S., Gupta, D. K., Santos, A. B., Cheng, S., Shah, A. M., et al. (2014). Sex-specific cardiovascular structure and function in heart failure with preserved ejection fraction. European Journal of Heart Failure, 16(5), 535–542. https://doi.org/10.1002/ejhf.67.
Zhang, M., Wu, J., Sun, R., Tao, X., Wang, X., Kang, Q., et al. (2019). SIRT5 deficiency suppresses mitochondrial ATP production and promotes AMPK activation in response to energy stress. PLoS One, 14(2), e0211796. https://doi.org/10.1371/journal.pone.0211796.
Douglas, P. S., Katz, S. E., Weinberg, E. O., Chen, M. H., Bishop, S. P., & Lorell, B. H. (1998). Hypertrophic remodeling: gender differences in the early response to left ventricular pressure overload. Journal of the American College of Cardiology, 32(4), 1118–1125.
Fliegner, D., Schubert, C., Penkalla, A., Witt, H., Kararigas, G., Dworatzek, E., et al. (2010). Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 298(6), R1597–R1606. https://doi.org/10.1152/ajpregu.00825.2009.
Ruppert, M., Korkmaz-Icoz, S., Loganathan, S., Jiang, W., Lehmann, L., Olah, A., et al. (2018). Pressure-volume analysis reveals characteristic sex-related differences in cardiac function in a rat model of aortic banding-induced myocardial hypertrophy. American Journal of Physiology. Heart and Circulatory Physiology, 315(3), H502–H511. https://doi.org/10.1152/ajpheart.00202.2018.
Previlon, M., Pezet, M., Vinet, L., Mercadier, J. J., & Rouet-Benzineb, P. (2014). Gender-specific potential inhibitory role of Ca2+/calmodulin dependent protein kinase phosphatase (CaMKP) in pressure-overloaded mouse heart. PLoS One, 9(6), e90822. https://doi.org/10.1371/journal.pone.0090822.
Scheuermann-Freestone, M., Freestone, N. S., Langenickel, T., Hohnel, K., Dietz, R., & Willenbrock, R. (2001). A new model of congestive heart failure in the mouse due to chronic volume overload. European Journal of Heart Failure, 3(5), 535–543.
Gardner, J. D., Brower, G. L., & Janicki, J. S. (2005). Effects of dietary phytoestrogens on cardiac remodeling secondary to chronic volume overload in female rats. Journal of Applied Physiology (Bethesda, MD: 1985), 99(4), 1378–1383. https://doi.org/10.1152/japplphysiol.01141.2004.
Dent, M. R., Tappia, P. S., & Dhalla, N. S. (2010). Gender differences in cardiac dysfunction and remodeling due to volume overload. Journal of Cardiac Failure, 16(5), 439–449. https://doi.org/10.1016/j.cardfail.2009.12.017.
Drolet, M. C., Lachance, D., Plante, E., Roussel, E., Couet, J., & Arsenault, M. (2006). Gender-related differences in left ventricular remodeling in chronic severe aortic valve regurgitation in rats. The Journal of Heart Valve Disease, 15(3), 345–351.
Beaumont, C., Walsh-Wilkinson, E., Drolet, M. C., Roussel, E., Arsenault, M., & Couet, J. (2017). Female rats with severe left ventricle volume overload exhibit more cardiac hypertrophy but fewer myocardial transcriptional changes than males. Scientific Reports, 7(1), 729. https://doi.org/10.1038/s41598-017-00855-9.
Dent, M. R., Tappia, P. S., & Dhalla, N. S. (2010). Gender differences in apoptotic signaling in heart failure due to volume overload. Apoptosis, 15(4), 499–510. https://doi.org/10.1007/s10495-009-0441-8.
Dent, M. R., Tappia, P. S., & Dhalla, N. S. (2011). Gender differences in beta-adrenoceptor system in cardiac hypertrophy due to arteriovenous fistula. Journal of Cellular Physiology, 226(1), 181–186. https://doi.org/10.1002/jcp.22321.
Wu, C. H., Liu, J. Y., Wu, J. P., Hsieh, Y. H., Liu, C. J., Hwang, J. M., et al. (2005). 17beta-estradiol reduces cardiac hypertrophy mediated through the up-regulation of PI3K/Akt and the suppression of calcineurin/NF-AT3 signaling pathways in rats. Life Sciences, 78(4), 347–356. https://doi.org/10.1016/j.lfs.2005.04.077.
Donaldson, C., Eder, S., Baker, C., Aronovitz, M. J., Weiss, A. D., Hall-Porter, M., et al. (2009). Estrogen attenuates left ventricular and cardiomyocyte hypertrophy by an estrogen receptor-dependent pathway that increases calcineurin degradation. Circulation Research, 104(2), 265–275, 211p following 275. https://doi.org/10.1161/circresaha.108.190397.
van Eickels, M., Grohe, C., Cleutjens, J. P., Janssen, B. J., Wellens, H. J., & Doevendans, P. A. (2001). 17beta-estradiol attenuates the development of pressure-overload hypertrophy. Circulation, 104(12), 1419–1423.
Iorga, A., Li, J., Sharma, S., Umar, S., Bopassa, J. C., Nadadur, R. D., et al. (2016). Rescue of Pressure Overload-Induced Heart Failure by Estrogen Therapy. Journal of the American Heart Association, 5(1). https://doi.org/10.1161/JAHA.115.002482.
Gardner, J. D., Murray, D. B., Voloshenyuk, T. G., Brower, G. L., Bradley, J. M., & Janicki, J. S. (2010). Estrogen attenuates chronic volume overload induced structural and functional remodeling in male rat hearts. American Journal of Physiology. Heart and Circulatory Physiology, 298(2), H497–H504. https://doi.org/10.1152/ajpheart.00336.2009.
Pedram, A., Razandi, M., Lubahn, D., Liu, J., Vannan, M., & Levin, E. R. (2008). Estrogen inhibits cardiac hypertrophy: role of estrogen receptor-beta to inhibit calcineurin. Endocrinology, 149(7), 3361–3369. https://doi.org/10.1210/en.2008-0133.
Kilic, A., Javadov, S., & Karmazyn, M. (2009). Estrogen exerts concentration-dependent pro-and anti-hypertrophic effects on adult cultured ventricular myocytes. Role of NHE-1 in estrogen-induced hypertrophy. Journal of Molecular and Cellular Cardiology, 46(3), 360–369. https://doi.org/10.1016/j.yjmcc.2008.11.018.
Gardner, J. D., Brower, G. L., Voloshenyuk, T. G., & Janicki, J. S. (2008). Cardioprotection in female rats subjected to chronic volume overload: synergistic interaction of estrogen and phytoestrogens. American Journal of Physiology. Heart and Circulatory Physiology, 294(1), H198–H204. https://doi.org/10.1152/ajpheart.00281.2007.
Li, Y., Kishimoto, I., Saito, Y., Harada, M., Kuwahara, K., Izumi, T., et al. (2004). Androgen contributes to gender-related cardiac hypertrophy and fibrosis in mice lacking the gene encoding guanylyl cyclase-A. Endocrinology, 145(2), 951–958. https://doi.org/10.1210/en.2003-0816.
Altamirano, F., Oyarce, C., Silva, P., Toyos, M., Wilson, C., Lavandero, S., et al. (2009). Testosterone induces cardiomyocyte hypertrophy through mammalian target of rapamycin complex 1 pathway. The Journal of Endocrinology, 202(2), 299–307. https://doi.org/10.1677/joe-09-0044.
Pirompol, P., Teekabut, V., Weerachatyanukul, W., Bupha-Intr, T., & Wattanapermpool, J. (2016). Supra-physiological dose of testosterone induces pathological cardiac hypertrophy. The Journal of Endocrinology, 229(1), 13–23. https://doi.org/10.1530/JOE-15-0506.
Duran, J., Oyarce, C., Pavez, M., Valladares, D., Basualto-Alarcon, C., Lagos, D., et al. (2016). GSK-3beta/NFAT Signaling Is Involved in Testosterone-Induced Cardiac Myocyte Hypertrophy. PLoS One, 11(12), e0168255. https://doi.org/10.1371/journal.pone.0168255.
Duran, J., Lagos, D., Pavez, M., Troncoso, M. F., Ramos, S., Barrientos, G., et al. (2017). Ca(2+)/Calmodulin-Dependent Protein Kinase II and Androgen Signaling Pathways Modulate MEF2 Activity in Testosterone-Induced Cardiac Myocyte Hypertrophy. Frontiers in Pharmacology, 8, 604. https://doi.org/10.3389/fphar.2017.00604.
Mishra, J. S., More, A. S., Gopalakrishnan, K., & Kumar, S. (2019). Testosterone plays a permissive role in angiotensin II-induced hypertension and cardiac hypertrophy in male rats. Biology of Reproduction, 100(1), 139–148. https://doi.org/10.1093/biolre/ioy179.
Regitz-Zagrosek, V., Oertelt-Prigione, S., Seeland, U., & Hetzer, R. (2010). Sex and gender differences in myocardial hypertrophy and heart failure. Circulation Journal, 74(7), 1265–1273.
de Kat, A. C., Dam, V., Onland-Moret, N. C., Eijkemans, M. J., Broekmans, F. J., & van der Schouw, Y. T. (2017). Unraveling the associations of age and menopause with cardiovascular risk factors in a large population-based study. BMC Medicine, 15(1), 2. https://doi.org/10.1186/s12916-016-0762-8.
Rossouw, J. E., Prentice, R. L., Manson, J. E., Wu, L., Barad, D., Barnabei, V. M., et al. (2007). Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA, 297(13), 1465–1477. https://doi.org/10.1001/jama.297.13.1465.
Miya, Y., Sumino, H., Ichikawa, S., Nakamura, T., Kanda, T., Kumakura, H., et al. (2002). Effects of hormone replacement therapy on left ventricular hypertrophy and growth-promoting factors in hypertensive postmenopausal women. Hypertension Research, 25(2), 153–159.
Dash, R., Schmidt, A. G., Pathak, A., Gerst, M. J., Biniakiewicz, D., Kadambi, V. J., et al. (2003). Differential regulation of p38 mitogen-activated protein kinase mediates gender-dependent catecholamine-induced hypertrophy. Cardiovascular Research, 57(3), 704–714. https://doi.org/10.1016/s0008-6363(02)00772-1.
Shen, T., Ding, L., Ruan, Y., Qin, W., Lin, Y., Xi, C., et al. (2014). SIRT1 functions as an important regulator of estrogen-mediated cardiomyocyte protection in angiotensin II-induced heart hypertrophy. Oxidative Medicine and Cellular Longevity, 2014, 713894. https://doi.org/10.1155/2014/713894.
Bhuiyan, M. S., Shioda, N., & Fukunaga, K. (2007). Ovariectomy augments pressure overload-induced hypertrophy associated with changes in Akt and nitric oxide synthase signaling pathways in female rats. American Journal of Physiology. Endocrinology and Metabolism, 293(6), E1606–E1614. https://doi.org/10.1152/ajpendo.00246.2007.
Sasaki, H., Nagayama, T., Blanton, R. M., Seo, K., Zhang, M., Zhu, G., et al. (2014). PDE5 inhibitor efficacy is estrogen dependent in female heart disease. The Journal of Clinical Investigation, 124(6), 2464–2471. https://doi.org/10.1172/JCI70731.
Redfield, M. M., Chen, H. H., Borlaug, B. A., Semigran, M. J., Lee, K. L., Lewis, G., et al. (2013). Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA, 309(12), 1268–1277. https://doi.org/10.1001/jama.2013.2024.
Zou, Y., Liang, Y., Gong, H., Zhou, N., Ma, H., Guan, A., et al. (2011). Ryanodine receptor type 2 is required for the development of pressure overload-induced cardiac hypertrophy. Hypertension, 58(6), 1099–1110. https://doi.org/10.1161/HYPERTENSIONAHA.111.173500.
Johnson, B. D., Zheng, W., Korach, K. S., Scheuer, T., Catterall, W. A., & Rubanyi, G. M. (1997). Increased expression of the cardiac L-type calcium channel in estrogen receptor-deficient mice. The Journal of General Physiology, 110(2), 135–140.
Jovanovic, S., Jovanovic, A., Shen, W. K., & Terzic, A. (2000). Low concentrations of 17beta-estradiol protect single cardiac cells against metabolic stress-induced Ca2+ loading. Journal of the American College of Cardiology, 36(3), 948–952.
Skavdahl, M., Steenbergen, C., Clark, J., Myers, P., Demianenko, T., Mao, L., et al. (2005). Estrogen receptor-beta mediates male-female differences in the development of pressure overload hypertrophy. American Journal of Physiology. Heart and Circulatory Physiology, 288(2), H469–H476. https://doi.org/10.1152/ajpheart.00723.2004.
Babiker, F. A., Lips, D., Meyer, R., Delvaux, E., Zandberg, P., Janssen, B., et al. (2006). Estrogen receptor beta protects the murine heart against left ventricular hypertrophy. Arteriosclerosis, Thrombosis, and Vascular Biology, 26(7), 1524–1530. https://doi.org/10.1161/01.atv.0000223344.11128.23.
Cavasin, M. A., Sankey, S. S., Yu, A. L., Menon, S., & Yang, X. P. (2003). Estrogen and testosterone have opposing effects on chronic cardiac remodeling and function in mice with myocardial infarction. American Journal of Physiology. Heart and Circulatory Physiology, 284(5), H1560–H1569. https://doi.org/10.1152/ajpheart.01087.2002.
Zwadlo, C., Schmidtmann, E., Szaroszyk, M., Kattih, B., Froese, N., Hinz, H., et al. (2015). Antiandrogenic therapy with finasteride attenuates cardiac hypertrophy and left ventricular dysfunction. Circulation, 131(12), 1071–1081. https://doi.org/10.1161/circulationaha.114.012066.
Baltatu, O., Cayla, C., Iliescu, R., Andreev, D., & Bader, M. (2003). Abolition of end-organ damage by antiandrogen treatment in female hypertensive transgenic rats. Hypertension, 41(3 Pt 2), 830–833. https://doi.org/10.1161/01.hyp.0000048702.55183.89.
Ziemens, B., Wallaschofski, H., Volzke, H., Rettig, R., Dorr, M., Nauck, M., et al. (2013). Positive association between testosterone, blood pressure, and hypertension in women: longitudinal findings from the Study of Health in Pomerania. Journal of Hypertension, 31(6), 1106–1113. https://doi.org/10.1097/HJH.0b013e3283603eb1.
Zhabyeyev, P., Gheblawi, M., & Oudit, G. Y. (2019). Testosterone and Cardiac Remodeling: Why are Older Men Susceptible to Heart Disease? American Journal of Physiology. Heart and Circulatory Physiology. https://doi.org/10.1152/ajpheart.00046.2019.
Rhoden, E. L., & Morgentaler, A. (2004). Risks of testosterone-replacement therapy and recommendations for monitoring. The New England Journal of Medicine, 350(5), 482–492. https://doi.org/10.1056/NEJMra022251.
Hwang, K., & Miner, M. (2015). Controversies in testosterone replacement therapy: testosterone and cardiovascular disease. Asian Journal of Andrology, 17(2), 187–191. https://doi.org/10.4103/1008-682X.146968.
Goodale, T., Sadhu, A., Petak, S., & Robbins, R. (2017). Testosterone and the heart. Methodist DeBakey Cardiovascular Journal, 13(2), 68–72. https://doi.org/10.14797/mdcj-13-2-68.
Ayaz, O., Banga, S. E., Heinze-Milne, S., Rose, R. A., Pyle, W. G., & Howlett, S. E. (2019). Long term testosterone deficiency modifies myofilament and calcium handling proteins and promotes diastolic dysfunction in the aging mouse heart. American Journal of Physiology. Heart and Circulatory Physiology. https://doi.org/10.1152/ajpheart.00471.2018.
Ikeda, Y., Aihara, K., Sato, T., Akaike, M., Yoshizumi, M., Suzaki, Y., et al. (2005). Androgen receptor gene knockout male mice exhibit impaired cardiac growth and exacerbation of angiotensin II-induced cardiac fibrosis. The Journal of Biological Chemistry, 280(33), 29661–29666. https://doi.org/10.1074/jbc.M411694200.
Ventura-Clapier, R., Dworatzek, E., Seeland, U., Kararigas, G., Arnal, J. F., Brunelleschi, S., et al. (2017). Sex in basic research: concepts in the cardiovascular field. Cardiovascular Research, 113(7), 711–724. https://doi.org/10.1093/cvr/cvx066.
Rohini, A., Agrawal, N., Koyani, C. N., & Singh, R. (2010). Molecular targets and regulators of cardiac hypertrophy. Pharmacological Research, 61(4), 269–280. https://doi.org/10.1016/j.phrs.2009.11.012.
de Simone, G., Devereux, R. B., Daniels, S. R., & Meyer, R. A. (1995). Gender differences in left ventricular growth. Hypertension, 26(6 Pt 1), 979–983.
Luczak, E. D., & Leinwand, L. A. (2009). Sex-based cardiac physiology. Annual Review of Physiology, 71, 1–18. https://doi.org/10.1146/annurev.physiol.010908.163156.
Grandi, A. M., Venco, A., Barzizza, F., Scalise, F., Pantaleo, P., & Finardi, G. (1992). Influence of age and sex on left ventricular anatomy and function in normals. Cardiology, 81(1), 8–13. https://doi.org/10.1159/000175770.
Sullivan, M. J., Cobb, F. R., & Higginbotham, M. B. (1991). Stroke volume increases by similar mechanisms during upright exercise in normal men and women. The American Journal of Cardiology, 67(16), 1405–1412.
Petersen, S. E., Hudsmith, L. E., Robson, M. D., Doll, H. A., Francis, J. M., Wiesmann, F., et al. (2006). Sex-specific characteristics of cardiac function, geometry, and mass in young adult elite athletes. Journal of Magnetic Resonance Imaging, 24(2), 297–303. https://doi.org/10.1002/jmri.20633.
Higginbotham, M. B., Morris, K. G., Coleman, R. E., & Cobb, F. R. (1984). Sex-related differences in the normal cardiac response to upright exercise. Circulation, 70(3), 357–366.
Foryst-Ludwig, A., & Kintscher, U. (2013). Sex differences in exercise-induced cardiac hypertrophy. Pflügers Archiv, 465(5), 731–737. https://doi.org/10.1007/s00424-013-1225-0.
Gertz, E. W., Wisneski, J. A., Stanley, W. C., & Neese, R. A. (1988). Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. The Journal of Clinical Investigation, 82(6), 2017–2025. https://doi.org/10.1172/jci113822.
Dorn, G. W., 2nd. (2007). The fuzzy logic of physiological cardiac hypertrophy. Hypertension, 49(5), 962–970. https://doi.org/10.1161/hypertensionaha.106.079426.
Horton, T. J., Dow, S., Armstrong, M., & Donahoo, W. T. (2009). Greater systemic lipolysis in women compared with men during moderate-dose infusion of epinephrine and/or norepinephrine. Journal of Applied Physiology (Bethesda, MD: 1985), 107(1), 200–210. https://doi.org/10.1152/japplphysiol.90812.2008.
Carter, S. L., Rennie, C., & Tarnopolsky, M. A. (2001). Substrate utilization during endurance exercise in men and women after endurance training. American Journal of Physiology. Endocrinology and Metabolism, 280(6), E898–E907. https://doi.org/10.1152/ajpendo.2001.280.6.E898.
Soto, P. F., Herrero, P., Schechtman, K. B., Waggoner, A. D., Baumstark, J. M., Ehsani, A. A., et al. (2008). Exercise training impacts the myocardial metabolism of older individuals in a gender-specific manner. American Journal of Physiology. Heart and Circulatory Physiology, 295(2), H842–H850. https://doi.org/10.1152/ajpheart.91426.2007.
Vega, R. B., Konhilas, J. P., Kelly, D. P., & Leinwand, L. A. (2017). Molecular Mechanisms Underlying Cardiac Adaptation to Exercise. Cell Metabolism, 25(5), 1012–1026. https://doi.org/10.1016/j.cmet.2017.04.025.
Foryst-Ludwig, A., Kreissl, M. C., Sprang, C., Thalke, B., Bohm, C., Benz, V., et al. (2011). Sex differences in physiological cardiac hypertrophy are associated with exercise-mediated changes in energy substrate availability. American Journal of Physiology. Heart and Circulatory Physiology. https://doi.org/10.1152/ajpheart.01222.2010.
Schaible, T. F., & Scheuer, J. (1979). Effects of physical training by running or swimming on ventricular performance of rat hearts. Journal of Applied Physiology: Respiratory, Environmental and Exercise Physiology, 46(4), 854–860. https://doi.org/10.1152/jappl.1979.46.4.854.
Schaible, T. F., & Scheuer, J. (1981). Cardiac function in hypertrophied hearts from chronically exercised female rats. Journal of Applied Physiology: Respiratory, Environmental and Exercise Physiology, 50(6), 1140–1145. https://doi.org/10.1152/jappl.1981.50.6.1140.
Strom, C. C., Aplin, M., Ploug, T., Christoffersen, T. E., Langfort, J., Viese, M., et al. (2005). Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. The FEBS Journal, 272(11), 2684–2695. https://doi.org/10.1111/j.1742-4658.2005.04684.x.
Honda, S., Harada, N., Ito, S., Takagi, Y., & Maeda, S. (1998). Disruption of sexual behavior in male aromatase-deficient mice lacking exons 1 and 2 of the cyp19 gene. Biochemical and Biophysical Research Communications, 252(2), 445–449.
Haines, C. D., Harvey, P. A., & Leinwand, L. A. (2012). Estrogens mediate cardiac hypertrophy in a stimulus-dependent manner. Endocrinology, 153(9), 4480–4490. https://doi.org/10.1210/en.2012-1353.
Ewer, M. S., & Gluck, S. (2009). A woman's heart: the impact of adjuvant endocrine therapy on cardiovascular health. Cancer, 115(9), 1813–1826. https://doi.org/10.1002/cncr.24219.
Mahmoodzadeh, S., Eder, S., Nordmeyer, J., Ehler, E., Huber, O., Martus, P., et al. (2006). Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 20(7), 926–934.
Power, R. F., Mani, S. K., Codina, J., Conneely, O. M., & O'Malley, B. W. (1991). Dopaminergic and ligand-independent activation of steroid hormone receptors. Science, 254(5038), 1636–1639.
Lehman, J. J., & Kelly, D. P. (2002). Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart Failure Reviews, 7(2), 175–185.
Arany, Z., He, H., Lin, J., Hoyer, K., Handschin, C., Toka, O., et al. (2005). Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metabolism, 1(4), 259–271. https://doi.org/10.1016/j.cmet.2005.03.002.
Grohe, C., Kahlert, S., Lobbert, K., & Vetter, H. (1998). Expression of oestrogen receptor alpha and beta in rat heart: role of local oestrogen synthesis. The Journal of Endocrinology, 156(2), R1–R7.
Funding
This work was supported by National Natural Science Foundation of China (31430039, 81670228, and 81730009), Laboratory Animal Science Foundation of Shanghai Committee of Science and Technology (16140901100), Scientific Research Foundation of Shanghai Municipal Commission of Health (201640044), Foundation for Key Researcher of Zhongshan Hospital (2017ZSYXQN09).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Associate Editor Yihua Bei oversaw the review of this article
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, J., Dai, F., Li, C. et al. Gender Differences in Cardiac Hypertrophy. J. of Cardiovasc. Trans. Res. 13, 73–84 (2020). https://doi.org/10.1007/s12265-019-09907-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-019-09907-z