
Abstract
Autism spectrum disorder (ASD) is a highly heritable neurodevelopmental disorder characterized by deficits in social interactions and repetitive behaviors. Although hundreds of ASD risk genes, implicated in synaptic formation and transcriptional regulation, have been identified through human genetic studies, the East Asian ASD cohorts are still under-represented in genome-wide genetic studies. Here, we applied whole-exome sequencing to 369 ASD trios including probands and unaffected parents of Chinese origin. Using a joint-calling analytical pipeline based on GATK toolkits, we identified numerous de novo mutations including 55 high-impact variants and 165 moderate-impact variants, as well as de novo copy number variations containing known ASD-related genes. Importantly, combined with single-cell sequencing data from the developing human brain, we found that the expression of genes with de novo mutations was specifically enriched in the pre-, post-central gyrus (PRC, PC) and banks of the superior temporal (BST) regions in the human brain. By further analyzing the brain imaging data with ASD and healthy controls, we found that the gray volume of the right BST in ASD patients was significantly decreased compared to healthy controls, suggesting the potential structural deficits associated with ASD. Finally, we found a decrease in the seed-based functional connectivity between BST/PC/PRC and sensory areas, the insula, as well as the frontal lobes in ASD patients. This work indicated that combinatorial analysis with genome-wide screening, single-cell sequencing, and brain imaging data reveal the brain regions contributing to the etiology of ASD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The current prevalence of ASD has increased to ~1 in 44 children aged 8 years old in the USA, and males are four times more susceptible to ASD than females [1]. The epidemiology survey in China showed that the prevalence of ASD ranges from 0.2% to 0.4%, suggesting that the difference in geography and clinical diagnosis criteria may lead to disparate impacts on studies of ASD etiology [2]. Recently, tremendous efforts in ASD genetic studies using whole-exome and whole-genome sequencing have built up high-throughput assessment pipelines for protein-disrupting variants in large ASD cohorts, in which de novo single nucleotide variants (SNVs), insertions and deletions (INDELs), and copy number variants (CNVs), as well as rare inherited variants, are major contributors to the genetic risks of ASD [3,4,5,6]. Thus, it is critical to further classify genetic causes from accumulated ASD genetic studies in consideration of neurobiological evidence. The online Simons Foundation Autism Research Initiative (SFARI) gene database provides an important public resource for ASD risk genes, in which over one thousand ASD candidate genes have been prioritized with genetic and neurobiological evidence (Category S, 1, 2, 3) [7].
Although genomic information of large cohorts consisting of >40,000 ASD patients and unaffected parents has been collected, East Asian populations are still underrepresented groups [8]. Some genetic studies on Chinese ASD cohorts using targeted multiplex sequencing technology focused on a group of genes associated with neurodevelopmental disorders, which cannot yield comprehensive genome-wide information about ASD risk genes [9, 10]. Two available genetic studies on Chinese ASD cohorts using whole-genome sequencing methods included <40 trios, limiting the power of genomic sequencing [11, 12].
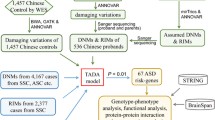
In this study, we applied whole-exome sequencing analysis to a Chinese cohort of 369 ASD probands with their parents. 150 bp paired-end sequencing short reads were mapped against the human reference genome build 38 (GRCh38/hg38). SNVs and INDELs were jointly called across all samples and filtered by GATK Variant Quality Score Recalibration (VQSR) and Convolutional Neural Network (CNN) tools. Together with analysis of single-cell sequencing data from the developing human brain, we found that the expression of genes with de novo mutations was specifically enriched in the pre-central and post-central gyrus (PRC and PC) and BST (banks of the superior temporal sulcus) in the human brain.
By further analysis of the brain imaging data with ASD and healthy controls, we found that the BST of the right hemisphere in ASD patients had a significantly decreased gray volume compared to healthy controls, suggesting potential structural deficits associated with the BST in autistic patients. Finally, after analyzing the seed-based functional connectivity (FC) of these regions, we found a decrease in FC between BST/PC/PRC and sensory areas, the insula, and the frontal lobes in ASD. This work indicated that the in-depth combinatorial analysis of ASD risk genes from genome-wide screening, single-cell sequencing, and brain imaging data can unveil the brain regions implicated in ASD and thus provide an analytical framework illustrating the genetic basis and neurobiological mechanism for ASD.
Materials and Methods
Samples and Ethics Statement
We analyzed a sample set consisting of 369 ASD probands and 706 parents from 353 pedigrees recruited from the Department of Child and Adolescent Psychiatry, Shanghai Mental Health Center. Of the families, 15 were multiplex that had two ASD children and 338 were trios. The fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) was used for ASD diagnoses made by trained psychiatrists. We obtained assent from the Institutional Review Board (IRB), Shanghai Mental Health Center of Shanghai Jiao Tong University (FWA number 00003065, IROG number 0002202). Dr. Yi-Feng Xu approved and signed our study with ethical review number 2016–4. Written informed consent was given by parents in consideration of the fact that all patients were minors. All participants were screened using the appropriate protocol approved by the IRB.
Whole-Exome Sequencing
Genomic DNA extracted from blood samples was sequenced at Shanghai Biotechnology Corporation\ and WuXi NextCODE on Illumina HiSeq sequencers using the Agilent SureSelect Human All Exon V5 exome capture kit. Some samples were sequenced at Euler Genomics on Illumina HiSeq sequencers using the IDT xGen Exome Research Panel v1 exome capture kit. 150-bp paired-end sequencing reads were aligned to human genome build 38 (GRCh38/hg38) using the Burrows-Wheeler Aligner [13], Picard tools MarkIlluminaAdapters, SamToFastq and MergeBamAlignment (http://broadinstitute.github.io/picard/) were aggregated into a BAM file. Per-individual coverages of the target regions calculated by Qualimap 2 are shown in Figure S1A [14]. The Picard tools MarkDuplicates, SortSam, and SetNmMdAndUqTags were used to mark duplicates, sort BAM files by chromosome coordinates, and add essential tags. SNVs and INDELs were jointly called across all samples using the Genome Analysis Toolkit (GATK) HaplotypeCaller 4.2.0.0 [15]. Variant call accuracy was estimated using the GATK VQSR approach and the GATK CNN Variant Filter. The VCF file (format v4.2) was produced by the Broad sequencing and calling pipeline with GATK version 4.2.0.0.
We included variant calls with the PASS flag in the downstream analyses. Variants (SNVs and INDELs) were annotated based on the hg38 database using VEP [16]. By following the definition of calculated variant consequences by VEP, we classified variants into those having HIGH, MODERATE, LOW, and MODIFIER impacts.
Population Stratification Using Genotyping Information of Frequent Exonic SNPs
To define a set of common exonic SNPs, we initially chose variants that were: (1) on the InfiniumExome-24v1-1_A1 genotyping array, (2) with MAF >0.05 in the East Asian (EAS) population of ExAC [17] annotated by VEP, and (3) biallelic in EAS. After combining the information of these SNPs in our cohort (OWN) with the data of the same SNPs in African (AFR), American (AMR), EAS, European (EUR), and South Asian (SAS) individuals in the 1000 Genomes Project [18], we applied further filtering and linkage disequilibrium (LD)-based pruning using PLINK v1.9 [19] with the following options and parameters: --maf (minor allele frequency) 0.05, --mind (maximum per-person missing) 0.2, --geno (maximum per-SNP missing) 0.2, --hwe (Hardy-Weinberg disequilibrium P-value) 1×10−10 and --indep (SNP window size, number of SNPs to shift and variance inflation factor threshold) [16, 19]. By using the data of 1064 SNPs that passed the filters described above, we applied multidimensional scaling with PLINK.
Identification of DNMs
We filtered out variant calls when one or more variant alleles occurred in unaffected parents of our cohort. By using the information of the remaining variant calls, we extracted candidates for DNMs using GATK PossibleDeNovo, TrioDenovo [20], and DeNovoGear [21]. Candidate DNMs called by these three tools at the same time were then stratified into SNVs and INDELs. We selected 161 DNM calls by prioritizing HIGH impact DNMs and 13 MODERATE impact DNMs. Possible-damaging missense DNMs were defined as the 14 variants predicted to be damaging by at least two of the seven prediction algorithms: SIFT [22], PolyPhen-2 HumVar [23], PolyPhen-2 HumDiv [23], LRT [24], MutationTaster [25], Mutation Assessor [26] and PROVEAN [27] annotated by dbNSFP4.0a [28, 29].
Results
Identification of de novo Variant in ASD Probands
We analyzed an ASD cohort consisting of 369 ASD probands and 706 parents from 353 pedigrees recruited from the Department of Child and Adolescent Psychiatry, Shanghai Mental Health Center. Among the cohort, there were 15 multiplex families containing two ASD children and 338 simplex families which had one ASD child. DSM-IV was used for ASD diagnoses by trained psychiatrists.
The proportion of the targeted exome regions covered by ≥20× or 40× of reads indicated sufficient coverage (Fig. S1A). After multidimensional scaling of the genotyping data, common exonic SNPs were identified with the PLINK toolkit (a whole genome association analysis tool) [30]. We found that common exonic SNPs in probands of the SMHC cohort were adjacent to the cluster of East Asian populations previously characterized, suggesting that the SMHC cohort faithfully carried the genetic signatures of East Asian populations (Fig. 1A).

Identification of De Novo Mutations in ASD Probands. A Multidimensional scaling plot of 369 ASD probands in our cohort (SMHC) along with African (AFR), American (AMR), East Asian (EAS), European (EUR), and South Asian (SAS) individuals in the 1000 Genomes Project. The figure was generated by analyzing genotyping data of 1,064 common exonic SNPs using PLINK. The first and second dimensions are shown. The orange, sky blue, bluish-green, yellow, and blue dots indicate AFR, AMR, EAS, and EUR SAS individuals. The vermilion dots indicate the ASD proband exomes sequenced in this study. B The number of families with various types of de novo mutations in our ASD cohort. “Others” indicates no detection of High- or Moderate-impact de novo mutations. C List of High-impact and Moderate-impact mutations identified in this ASD cohort. The genes also present in the SFARI gene list are highlighted with various colors representing 4 categories (Cat S, 1–3).
After variant filtering, we discovered a set of 220 de novo mutations (DNMs) (Table S1). We classified DNMs into three categories, High-impact, Moderate-impact, and Possible-damaging. The High- and Moderate-impact categories were defined by VEP (Ensembl Variant Effect Predictor, https://asia.ensembl.org/info/docs/tools/vep/index.html). Briefly, the High-impact variants usually led to the truncation of protein products, including the gain or loss of STOP codons as well as frameshift-causing INDELs. Interestingly, among the 55 genes containing High-impact SNVs, there were 18 genes previously reported in the SFARI gene list (Category S, 1, 2, 3) such as SCN2A, PTEN, MECP2, SRCAP, and TCF4, indicating that most genes containing high-impact SNVs in the Chinese cohorts are novel and not included in the SFARI gene database. (Fig. 1B, C).
Moderate-impact variants were defined as changing, but not truncating the protein sequence, such as missense SNVs and inframe INDELs. To further categorize the severity of missense variants, we annotated missense SNVs into a new class, named Possible-damaging missense DNMs, which were defined as the variants predicted to be damaging by at least two of the seven following prediction algorithms: SIFT [22], PolyPhen-2 HumVar [23], PolyPhen-2 HumDiv [23], LRT [24], Mutation Taster [25], Mutation Assessor [26] and PROVEAN [27] annotated by dbNSFP4.0a [28, 29]. Interestingly, among 165 Moderate-impact variants, only 23 variants were present in the SFARI gene list (Fig. 1B, C).
Over one thousand ASD risk genes in the SFARI gene list were mainly found from genetic studies in American and European studies, therefore we wondered whether numerous genes with DNMs in the Chinese ASD cohorts which were not included in the SFARI list really contributed to ASD or some common genetic variants may not be associated with disorders. To further determine whether these genes with DNMs may contribute to ASD, we next statistically evaluated the contributions of each de novo variant to ASD using the Transmission and De Novo Association Test-Denovo (TADA-Denovo) method. We first measured the frequency of de novo and missense variants in each gene with DNMR-SC-subtype data [31], then applied the TADA-Denovo method [32]. We classified the DNM variants with p values obtained from the TADA-Denovo test into two tiers (*P <0.01, or **P <0.001) (Table S2). We further measured the “probability of loss-of-function intolerance” (pLi) score for each variant and categorized variants with significant TADA-Denovo values into two tiers as well (0.9 represented by ## and 0.5–0.9 represented by #) [17]. Finally, we found that 11 genes with High-impact mutations and 35 genes with Moderate-impact mutations, none of which were included in the SFARI gene list, had statistically significant TADA-Denovo and pLi scores, further strengthening their contributions to ASD (Fig. 1C).
We then investigated whether genes with de novo variants identified in various ASD genetic studies overlap. Interestingly, we found that de novo ASD risk genes detected in ASD probands in the SMHC cohort showed little overlap with the list of de novo ASD risk genes from the Japanese cohort (Fig. S1B, C) [8]. Moreover, we found that there was also little overlap in de novo variants between the SMHC cohorts with other studies with 200–400 trios (Fig. S1D) [5, 33,34,35].
Identification of de novo CNVs in ASD Risk Genes with the WES Dataset
Although the gold standard for the detection of copy number variations is chromosomal microarray analysis, various toolkits have emerged to identify CNVs with whole-exome sequencing (WES) datasets [36]. However, the current algorithms for CNV detection are not optimal for the WES dataset and are incompatible with the GRCh38/hg38 reference genome.
We applied a germline CNV-calling protocol based on the GATK cohort mode (version 4.2.0.0) (see Supplementary Methods) and identified numerous de novo CNVs in the probands (Fig. 2A–N, Table S3). To exclude false-positive hits, we set 2 standards for CNV screening. First, the selection of duplication or deletion signals appearing in >2 continuous exons. Second, CNVs should fulfill the HIGH-impact criteria, leading to protein truncation, such as the deletion of START or STOP codons.

Identification of De Novo CNVs. A–N Schematics of 18 SFARI ASD risk genes (Cat S: 4 genes, Cat 1: 14 genes) with de novo CNVs (blue, deleted chromosomal segment; red, duplicated chromosomal segments). O Quantitative real-time PCR verification of de novo CNVs. TBR1, t-test, P = 3.22×10−7; RAI1, t-test, P = 7.40×10−7; MECP2, t-test, P = 0.0069; SHANK3, t-test, P = 0.0028, GIGYF1, t-test, P = 0.0335. All experiments were repeated >4 independent times. Error bars represent standard errors from 4 or 6 replicates.
To prioritize ASD risk genes, we first examined CNVs that occurred in the known SFARI genes (Fig. 2A–N). We found 18 CNVs exhibiting duplication or deletions in known SFARI genes (Cat S:4 genes, Cat 1:14 genes), such as duplications of RAI1 and UBE3A, and deletions of TBR1, SHANK3, MECP2, and GIGYF1 (Fig. 2A–N). We further validated the CNV results by using quantitative PCR, confirming the feasibility and faithfulness of our new methods (Fig. 2O).
Furthermore, among the de novo large CNVs we found, there were 9 CNVs containing genes in the SFARI Cat 2 gene list (Table S3). There were a total of 26 CNVs containing critical ASD-risk genes in the SFARI gene list (Cat S, 1, 2), suggesting that genes implicated in these de novo large CNVs contribute to the pathogenesis of ASD.
Expression of ASD Risk Genes Enriched in the PC, PRC, and BST Regions of the Developing Human Brain
The etiology of ASD may involve disruption of neural circuits associated with social behaviors, thus identification of the expression profile of genes with DNMs in the human brain would provide critical insights into which brain regions may be affected by mutations of ASD risk genes [37]. To acquire the expression pattern of ASD risk genes at single-cell resolution, we used the recent single-cell sequencing database in the developing human brain [38, 39]. We grouped a total of 17,434 transcriptomes of human fetal brains collected from gestational weeks 9 to 26 and categorized them into sub-cell types according to marker genes (Figs. 3A and S2A–C).

Expression pattern of ASD risk genes among different cell types. A Clustering of single-cell RNA-seq data from different brain regions. Cell types are colored differently. B Visualization of enrichment scores of the 55 High-impact genes by UMAP. C Visualization of enrichment scores of the 165 Moderate-impact genes by UMAP. D Heatmap showing averaged enrichment scores of ASD risk genes among the different subtypes (high, red; low, blue). The top three most enriched cell types for each gene set are shown on the sidebar. E Histogram showing the proportion of highly expressed genes (genes expressed in at least 25% of cells in an individual cell type) of the 55 High-impact genes among cell types. F Histogram showing the proportion of highly expressed genes (genes expressed in at least 25% of cells in an individual cell type) of the 165 Moderate-impact genes among cell types.
We first examined the expression pattern of 55 High-impact genes and 165 Moderate-impact genes in various cell types and found that both categories of genes were highly expressed in several subtypes of cells, including NPC-4, Ex-1, and In-2, as well as Cajal-Retzius (CR) cells (Fig. 3B–D). We further looked into where NPC-4, Ex-1, In-2, and handcar cells are localized in the developing human brain. We found that NPC-4 was generally distributed in the four major lobes of the brain, suggesting that this specific sub-group of neural progenitor cells may be associated with ASD (Figs. 3E, F and S3A, B). However, Ex-1 and In-2 were specifically enriched in some sub-regions of the brain including the PRC, PC, and BST regions (Fig. S3C–E).
We next investigated whether the expression of ASD risk genes is enriched in specific regions of the human brain. In previous work, single-cell sequencing was applied to 22 brain subregions in the developing human brain (Fig. 4A) [38]. Surprisingly, we found that the High- and Moderate-impact genes were significantly enriched in the PRC, PC, and BST regions (Fig. 4B–E). The PRC includes the primary motor cortex (M1), and the PC includes the primary somatosensory cortex (S1). The involvement of the PRC and PC in ASD had been reported previously [40, 41]. Interestingly, we also found that the FC including the right S1 and M1 regions is specifically decreased in MECP2 transgenic monkeys, the non-human primate model for autism, compared to wild-type monkeys [42,43,44].

Expression pattern of ASD risk genes among different brain regions. A Schematic showing the 22 brain sub-regions used in published human single-cell RNA-seq data and applied in the analysis of this work. B UMAP display of diverse brain regions (abbreviations are listed in Table S7). Brain sub-regions are differently colored. C Heatmap showing averaged enrichment scores of ASD risk genes among different brain regions (high, red; low, blue). The top three most enriched brain regions for each gene set are shown on the sidebar. D–E Histograms displaying the proportions of highly expressed genes (genes expressed in at least 25% of cells from individual brain regions) of the 55 High-impact genes (D) and the 165 Moderate-impact genes (E) among various brain sub-regions.
Brain Imaging Analysis
To determine whether these brain regions were affected in ASD patients from different populations, we acquired imaging data from the Autism Brain Imaging Data Exchange (ABIDE I, http://fcon_1000.projects.nitrc.org/indi/abide/) [45] a publicly available database containing 1112 subjects [539 ASDs, 573 age-matched healthy controls (HCs)] from 16 international imaging sites who underwent anatomical and resting-state functional MRI scans. We collected >200 age-matched brain imaging data from ASD or HC groups in ABIDE-I (Table S4).
To further test whether the PC/PRC and BST have structural (gray matter) alternations in ASD patients, we first applied voxel-based morphometry (VBM) analysis of these regions in ASD and HC using T1 data. Surprisingly, we found that the gray matter volume of the BST in the right hemisphere was significantly smaller in the ASD group than in the HC group (t = 3.61, P = 0.003, t- and P-values from the linear mixed model detailed in the Statistics section of Materials and Methods), and this effect persisted even after controlling for medication status (t = 3.32, P = 0.001) and full-scale intelligence quotient (FIQ) (t = 3.4, P = 0.0007) (Fig. 5A–C).

Differences in structural and functional connectivity between ASD and healthy controls in the PC, PRC, and BST regions. A VBM. Beta, t, P, and degrees of freedom (DF) are obtained from linear mixed models. Beta is the regression coefficient of the fixed effect. DF is the degree of freedom of the model (equal to the sample size minus the number of parameters to be estimated). T stands for t-statistics for testing the null hypothesis that the coefficient is equal to zero. P stands for the P-value for the t-test. B VBM data with control of the FIQ effect. C VMB data excluding subjects with medications. D Group differences between ASD and healthy controls in seed-based resting-state functional connectivity (FC). Six brain regions including the bilateral BST, PC, and PRC were selected as seeds. The FC between these 6 regions and the whole-brain voxels was calculated. Only the clusters of voxels with significant FC differences between the two groups (the two-sample t-test, PFWE <0.05, cluster size 20 voxels or 180 mm3) are displayed.
We finally investigated the potential FC between the above regions of interest (ROIs) and the whole-brain voxels, by applying seed-based FC analysis using resting-state functional MRI scan data from ASD and HCs. Consistently, we found a significant decrease in connectivity between the BST/PC/PRC and sensory areas, insula, and frontal lobes in ASD compared to HC (Fig. 5D). We found a decrease in the FC of all six ROIs in the occipital lobe, which is commonly associated with vision. We also found decreased connectivity between the bilateral PC/PRC to the sensorimotor region of the parietal lobe. In addition, in the right BST, we found the widest FC decrease among all ROIs, including connections to the right insula and temporal lobes (t = –6.05, FWE corrected P = 0.0002), to the bilateral frontal lobe and the occipital lobe (Table S5–S6 and Fig. 5D).
The BST includes voice-selective areas in normal adults and plays a role in voice recognition and processing of social stimuli [46]. In an fMRI study, activation of the BST by speech stimulation appeared to be compromised in adults with ASD [47]. In addition, the BST also exhibits ASD-related functional connectivity alterations [48, 49], gray matter changes (such as lower surface area and greater age-related cortical thinning) [50, 51], and white matter volume reduction [52]. Our study indicated that genetic predispositions in ASD patients may lead to structural and functional abnormalities in brain regions associated with the processing of social information, thus providing novel candidate brain regions for interventions in ASD.
Discussion
With accumulating genomic studies on autism cohorts worldwide, the genetic architecture of ASD has emerged over the last decade. Composed of de novo and rare inherited mutations, genetic variants play a decisive role in determining the etiology of ASD. Although the rapid development of DNA sequencing technology, precise identification of genetic variants in large-scale genome sequencing over hundreds and thousands of ASD core trios is still very challenging.
In this work, we applied the latest GATK package (v4.2.0.0) and the GRCh38/hg38 dataset, which is compatible with the ongoing update of the Ensembl genome database. We focused on the identification of de novo variants, including SNVs, INDELs, and CNVs, with a customized joint calling pipeline. Importantly, we found several critical CNVs containing ASD-risk genes, such as SHANK3, TBR1, and MECP2, indicating that screening CNVs with the WES dataset is very valuable for ASD genetic studies. Interestingly, ~30% (18/55) of genes carried de novo high-impact variants that were present in the SFARI gene list, suggesting that there are potentially novel ASD genes in the Chinese cohorts. With the in-depth analysis of TADA-Denovo and pLi evaluation, we found that a substantial portion of DNM discovered in the Chinese ASD cohorts was significant statistically. Taken together, we suggest that although the overall genetic architecture of ASD remains similar across different populations, the frequency of individual genetic components may vary due to geographic isolation. Thus, to comprehensively acquire the ASD risk genes, genome-wide sequencing in large cohorts from different populations is required.
Further in-depth analysis will require the correlation between genetic variants with behavioral abnormalities and cognitive features including IQ. ASD patients are usually associated with various degrees of repetitive behaviors and developmental delay. It would be valuable to precisely classify the genetic variants with behavioral features and developmental stages. In the current cohorts, all ASD patients were diagnosed based on DSM-IV but lacked further cognitive examinations. Thus whether de novo variants are associated with particular behavioral symptoms remains to be addressed.
One intriguing hypothesis of the etiology of ASD is that genes carrying mutations in ASD patients may be expressed in specific brain regions governing social behavior-related circuits. Thus precise identification of the expression pattern of ASD risk genes in the human brain, with single-cell resolution, would be an ideal approach. In this work, we took advantage of a single-cell sequencing database collected from various brain regions across gestational weeks 9 to 26 and found that the expression profiles of ASD risk genes in the developing human brain indeed exhibit specific patterns. ASD risk genes discovered in the Chinese ASD cohort were specifically enriched in the primary somatosensory (S1-PC) and primary motor cortices (M1-PRC), as well as the BST region.
Although there is evidence suggesting that the S1 and M1 cortices show defects in functional connectivity in ASD samples [42,43,44], it is still difficult to determine whether these defects are the cause or consequence of deficits in social behaviors. It is well known that ASD patients often exhibit abnormal somatosensory functions. Thus our data suggested that abnormalities in somatosensory functions may be rooted in central control, rather than peripheral functions.
The finding that the right BST region is implicated in ASD pathogenesis is extremely intriguing. That the regions in the right hemisphere associated with autism are stronger than those in the left hemisphere has also been reported in previous work [42,43,44]. The connection of the right hemisphere with autism has also been discussed [53]. The involvement of the BST in social perception has been found in previous work with human subjects [54]. Thus we hypothesized that dysregulation of ASD risk genes in the BST likely causes abnormalities in social perception and related functions. Since the BST is on the surface of the human brain, one may be able to design neuromodulation methods to activate neural activity in the right BST region of ASD patients, through transcranial electrical or magnetic stimulation.
Taken together, this work presents numerous ASD risk genes from the whole-exome sequencing of 369 Chinese ASD cases. Besides known ASD candidate genes present in the SFARI gene database, we further measured the probability of DNM contributions to ASD through the TADA-Denovo test and found that quite a few ASD candidate genes appeared to be statistically significant, suggesting that whole-exome sequencing on large ASD cohorts are indeed valuable to elucidate the genetic landscape of ASD in different populations. Importantly, the combinational analysis of single-cell sequencing and brain imaging in this work presented an analytical framework in which one could address the potential etiology of ASD from genetic discoveries.
Data and Materials Availability
The datasets used and/or analyzed during the current study are available from the lead contact on reasonable request.
References
Shaw KA, Maenner MJ, Bakian AV, Bilder DA, Durkin MS, Furnier SM. Early identification of autism spectrum disorder among children aged 4 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill Summ 2021, 70: 1–14.
Zhou H, Xu X, Yan W, Zou X, Wu L, Luo X, et al. Prevalence of autism spectrum disorder in China: A nationwide multi-center population-based study among children aged 6 to 12 years. Neurosci Bull 2020, 36: 961–971.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515: 209–215.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515: 216–221.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485: 237–241.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020, 180: 568-584.e23.
Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, et al. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism 2013, 4: 36.
Takata A, Miyake N, Tsurusaki Y, Fukai R, Miyatake S, Koshimizu E, et al. Integrative analyses of de novo mutations provide deeper biological insights into autism spectrum disorder. Cell Rep 2018, 22: 734–747.
Guo H, Wang T, Wu H, Long M, Coe BP, Li H, et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol Autism 2018, 9: 64.
Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 2016, 7: 13316.
Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet 2013, 93: 249–263.
Wu J, Yu P, Jin X, Xu X, Li J, Li Z, et al. Genomic landscapes of Chinese sporadic autism spectrum disorders revealed by whole-genome sequencing. J Genet Genomics 2018, 45: 527–538.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25: 1754–1760.
Okonechnikov K, Conesa A, García-Alcalde F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32: 292–294.
Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018, https://doi.org/10.1101/201178.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol 2016, 17: 122.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536: 285–291.
1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1, 092 human genomes. Nature 2012, 491: 56–65.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4: 7.
Wei Q, Zhan X, Zhong X, Liu Y, Han Y, Chen W, et al. A Bayesian framework for de novo mutation calling in parents-offspring trios. Bioinformatics 2015, 31: 1375–1381.
Ramu A, Noordam MJ, Schwartz RS, Wuster A, Hurles ME, Cartwright RA, et al. DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods 2013, 10: 985–987.
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc 2016, 11: 1–9.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010, 7: 248–249.
Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res 2009, 19: 1553–1561.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010, 7: 575–576.
Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res 2011, 39: e118.
Choi Y, Chan AP. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31: 2745–2747.
Liu X, Jian X, Boerwinkle E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat 2011, 32: 894–899.
Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 2016, 37: 235–241.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007, 81: 559–575.
Jiang Y, Li Z, Liu Z, Chen D, Wu W, Du Y, et al. mirDNMR: A gene-centered database of background de novo mutation rates in human. Nucleic Acids Res 2017, 45: D796–D803.
He X, Sanders SJ, Liu L, de Rubeis S, Lim ET, Sutcliffe JS, et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet 2013, 9: e1003671.
Feliciano P, Zhou X, Astrovskaya I, Turner TN, Wang T, Brueggeman L, et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom Med 2019, 4: 19.
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485: 242–245.
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485: 246–250.
Enomoto Y, Tsurusaki Y, Yokoi T, Abe-Hatano C, Ida K, Naruto T, et al. CNV analysis using whole exome sequencing identified biallelic CNVs of VPS13B in siblings with intellectual disability. Eur J Med Genet 2020, 63: 103610.
Geschwind DH, State MW. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol 2015, 14: 1109–1120.
Fan X, Dong J, Zhong S, Wei Y, Wu Q, Yan L, et al. Spatial transcriptomic survey of human embryonic cerebral cortex by single-cell RNA-seq analysis. Cell Res 2018, 28: 730–745.
Fan X, Fu Y, Zhou X, Sun L, Yang M, Wang M, et al. Single-cell transcriptome analysis reveals cell lineage specification in temporal-spatial patterns in human cortical development. Sci Adv 2020, 6: eaaz2978.
Ayub R, Sun KL, Flores RE, Lam VT, Jo B, Saggar M, et al. Thalamocortical connectivity is associated with autism symptoms in high-functioning adults with autism and typically developing adults. Transl Psychiatry 2021, 11: 93.
Mizuno Y, Kagitani-Shimono K, Jung M, Makita K, Takiguchi S, Fujisawa TX, et al. Structural brain abnormalities in children and adolescents with comorbid autism spectrum disorder and attention-deficit/hyperactivity disorder. Transl Psychiatry 2019, 9: 332.
Cai DC, Wang Z, Bo T, Yan S, Liu Y, Liu Z, et al. MECP2 duplication causes aberrant GABA pathways, circuits and behaviors in transgenic monkeys: Neural mappings to patients with autism. J Neurosci 2020, 40: 3799–3814.
Liu Z, Li X, Zhang JT, Cai YJ, Cheng TL, Cheng C, et al. Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 2016, 530: 98–102.
Zhan Y, Wei J, Liang J, Xu X, He R, Robbins TW, et al. Diagnostic classification for human autism and obsessive-compulsive disorder based on machine learning from a primate genetic model. Am J Psychiatry 2021, 178: 65–76.
Di Martino A, Yan CG, Li Q, Denio E, Castellanos FX, Alaerts K, et al. The autism brain imaging data exchange: Towards a large-scale evaluation of the intrinsic brain architecture in autism. Mol Psychiatry 2014, 19: 659–667.
Belin P, Zatorre RJ, Lafaille P, Ahad P, Pike B. Voice-selective areas in human auditory cortex. Nature 2000, 403: 309–312.
Gervais H, Belin P, Boddaert N, Leboyer M, Coez A, Sfaello I, et al. Abnormal cortical voice processing in autism. Nat Neurosci 2004, 7: 801–802.
Shih P, Keehn B, Oram JK, Leyden KM, Keown CL, Müller RA. Functional differentiation of posterior superior temporal sulcus in autism: A functional connectivity magnetic resonance imaging study. Biol Psychiatry 2011, 70: 270–277.
Venkataraman A, Duncan JS, Yang DY, Pelphrey KA. An unbiased Bayesian approach to functional connectomics implicates social-communication networks in autism. Neuroimage Clin 2015, 8: 356–366.
Mensen VT, Wierenga LM, van Dijk S, Rijks Y, Oranje B, Mandl RC, et al. Development of cortical thickness and surface area in autism spectrum disorder. Neuroimage Clin 2017, 13: 215–222.
Braden BB, Riecken C. Thinning faster? Age-related cortical thickness differences in adults with autism spectrum disorder. Res Autism Spectr Disord 2019, 64: 31–38.
von dem Hagen EA, Nummenmaa L, Yu R, Engell AD, Ewbank MP, Calder AJ. Autism spectrum traits in the typical population predict structure and function in the posterior superior temporal sulcus. Cereb Cortex 2011, 21: 493–500.
Ozonoff S, Miller JN. An exploration of right-hemisphere contributions to the pragmatic impairments of autism. Brain Lang 1996, 52: 411–434.
Deen B, Koldewyn K, Kanwisher N, Saxe R. Functional organization of social perception and cognition in the superior temporal sulcus. Cereb Cortex 2015, 25: 4596–4609.
Acknowledgements
We thank the families for their participation in this study. This work was supported by the National Natural Science Foundation of China (31625013, 81941015, 32000726, and 61973086), the Shanghai Brain-Intelligence Project from STCSM (16JC1420501), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDBS01060200), the Program of Shanghai Academic Research Leader, The Open Large Infrastructure Research of the Chinese Academy of Sciences, and the Shanghai Municipal Science and Technology Major Project (2018SHZDZX01).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yuan, B., Wang, M., Wu, X. et al. Identification of de novo Mutations in the Chinese Autism Spectrum Disorder Cohort via Whole-Exome Sequencing Unveils Brain Regions Implicated in Autism. Neurosci. Bull. 39, 1469–1480 (2023). https://doi.org/10.1007/s12264-023-01037-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-023-01037-6