
Abstract
Since the discovery of the C9ORF72 gene in 2011, great advances have been achieved in its genetics and in identifying its role in disease models and pathological mechanisms; it is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). ALS patients with C9ORF72 expansion show heterogeneous symptoms. Those who are C9ORF72 expansion carriers have shorter survival after disease onset than non-C9ORF72 expansion patients. Pathological and clinical features of C9ORF72 patients have been well mimicked via several models, including induced pluripotent stem cell-derived neurons and transgenic mice that were embedded with bacterial artificial chromosome construct and that overexpressing dipeptide repeat proteins. The mechanisms implicated in C9ORF72 pathology include DNA damage, changes of RNA metabolism, alteration of phase separation, and impairment of nucleocytoplasmic transport, which may underlie C9ORF72 expansion-related ALS/FTD and provide insight into non-C9ORF72 expansion-related ALS, FTD, and other neurodegenerative diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by a deficiency of upper and lower motor neurons in the motor cortex and lumbar spinal cord, respectively [1]. Frontotemporal dementia (FTD) is a devastating disease that mainly involves the frontal and temporal lobes [2]. A GGGGCC (G4C2) hexanucleotide repeat expansion (HRE) in the intron of the C9ORF72 gene was identified in 2011 and is the most common genetic cause of both ALS and FTD [3,4,5]. Tens to thousands of G4C2 repeats have been identified in carriers and patients with the C9ORF72-related ALS and FTD (c9ALS/FTD) mutation, while only ~30 repeats occur in normal individuals [3, 4]. The HRE can be further transcribed and translated into sense and antisense RNAs, as well as dipeptide repeat proteins (DPRs) that include poly-GA, poly-GP, poly-GR, poly-PA, and poly-PR [6,7,8,9,10,11,12].
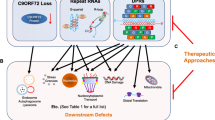
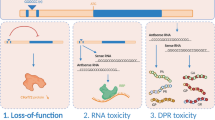
The underlying mechanisms of c9ALS/FTD can be classified into three prototypes (Fig. 1): (1) loss-of-function of the C9ORF72 protein [13,14,15,16,17]; (2) formation of sense and antisense RNA foci in the nucleus [18,19,20]; and (3) gain-of-function caused by repeat-associated non-ATG-initiated translation of DPRs [21,22,23,24]. In recent years, several studies have implicated C9ORF72 in cellular protein transport and that loss of C9ORF72 impairs autophagy [13, 14, 25,26,27] and lysosome biogenesis [28]. Despite the fact that C9ORF72 loss-of-function contributes to microglial activation and a “cytokine storm” in several transgenic mouse models, reducing the expression of C9ORF72 alone does not induce c9ALS/FTD phenotypes and is dispensable for neuronal survival [15,16,17, 26, 29]. Thus, we summarize the literature focusing on gain-of-function in this review.

Mechanisms underlying the GGGGCC hexanucleotide repeat expansion. (1) Loss-of-function: Decreased expression of C9orf72 mRNA in the frontal cortex of individuals with C9ORF72 mutation. (2) Gain-of-function caused by RNA foci: The GGGGCC repeat expansion is transcribed into repeat RNA that can interact with DNA to fold into a G-quadruplex structure. The repeat RNA forms RNA foci that sequester RNA-binding proteins in the nucleus of vulnerable neurons. (3) Gain-of-function caused by dipeptide repeat proteins (DPRs): In a repeat-associated non-ATG-initiated manner, the repeat RNA is translated into DPRs that form toxic aggregates in residual neurons.
Clinical Features
Genetics
The G4C2 HRE in the noncoding region of the C9ORF72 gene contributes to ~25.1% of familial FTD and 37.6% of familial ALS world-wide [30,31,32]. Strikingly, in the Finnish population, C9ORF72 repeat expansion accounts for up to 46.0% of familial ALS and 21.1% of sporadic ALS [4], but the frequency of C9ORF72 repeat expansion is extremely low in Asian populations [33,34,35]. In recent years, several studies have comprehensively described the clinical and pathological features of C9ORF72 patients who exhibit wide variation of age at onset and disease duration [36,37,38,39,40,41,42,43,44]. Most bulbar-onset C9ORF72 patients exhibit symptomatic heterogeneity [38, 39, 43], including behavioral-variant FTD, progressive non-fluent aphasia, motor neuron disease, ALS, and mild psychosis and anxiety symptoms [41,42,43]. The ALS patients with C9ORF72 expansion have shorter survival after disease onset than those without C9ORF72 expansion [45].
Pathological Features
Neuronal Deficiency
Within a cohort of patients with behavioral variant FTD, compared with patients with mutation in microtubule-associated protein tau (MAPT) and progranulin (GRN) that display specific anteromedial temporal atrophy [46, 47] and temporoparietal atrophy [48, 49], respectively, the patients with HRE in C9ORF72 show a widespread pattern of grey matter loss in the cerebellum and spinal cord, and the extramotor frontal lobes, temporal lobes, and hippocampus, as well as the basal ganglia and occipital lobes [37,38,39,40,41,42,43,44, 50]. Notably, an atrophied cerebellum, which is a characteristic pathology of c9ALS/FTD, is not presented in brain of FTD cases without C9ORF72 expansion [51, 52], suggesting a specific role of C9ORF72 in cerebellar pathogenesis. Several other studies using magnetic resonance imaging (MRI) and unbiased voxel-based morphometric analysis have also found broad brain atrophy in C9ORF72 patients [41, 44], thereby accounting for the clinical heterogeneity of these patients.
Pathological Inclusions
One specific pathological feature of c9ALS/FTD is the presence of G4C2 nuclear RNA foci in both sense and antisense forms [3, 8]; the other is cytoplasmic inclusions of RNA-translated DPRs [9,10,11,12, 53]. Notably, intranuclear inclusions of DPRs that co-localize with nucleoli have been identified in the frontal cortex of C9ORF72 patients [54, 55]. The expression of the five DPRs is predominantly in the form of poly-GA, to a lesser extent poly-GP and poly-GR, compared to poly-PA and poly-PR [53]. However, only slight clinico-pathological correlations of poly-GA, but not other DPRs, have been reported in c9ALS/FTD [53]. It is still unclear how much other DPRs contribute to the pathogenesis, especially arginine-rich poly-GR/PR.
TDP-43 is a major component in the pathological inclusions in the ALS and FTD brain [56,57,58]. FTD-TDP is classified into four types according to the heterogeneity of TDP-43 inclusions: cytoplasmic inclusions, short neurites in the upper cortical layers (Type A), round TDP-43 inclusions throughout the cortex (Type B), long dystrophic neurites (Type C), and intranuclear inclusions (Type D) [59]. Broad inclusions of TDP-43 have also been identified in the vulnerable neurons of c9ALS/FTD, mainly containing type A, type B, or a combination of both [59,60,61,62]. A series of studies have reported that p62-positive, TDP-43-negative cytoplasmic inclusions are present in the cerebellar granular cells of c9ALS/FTD patients, while such inclusions are not found in non-C9ORF72 mutant individuals [40, 42, 53, 61, 63, 64], indicating a specific role of HRE in the protein degradation pathway. The clinical and pathological features of c9ALS/FTD described elsewhere are summarized in Table 1.
In recent years, impaired nucleocytoplasmic transport has been identified as a common pathological process in C9ORF72 expansion-induced neurodegeneration [65,66,67,68]. Using large-scale unbiased genetic screening, several genes in the nucleocytoplasmic transport process have been identified as major hints in G4C2-expressing Drosophila models [66]. Consistently, RanGAP1, a key regulator of nucleocytoplasmic transport [69, 70], is abnormally distributed in the cortex of the G4C2 mouse model and C9ORF72 ALS patients [67].
Mouse Models with Hexanucleotide Repeat Expansion
It has been reported that overexpression of HRE causes obvious cellular toxicity in cell cultures [18, 19, 23], G4C2 Drosophila models [71,72,73], and a G4C2 zebrafish model [20]. In addition, Petrucelli and colleagues have developed two G4C2-expressing mouse models using an AAV-packaged hexanucleotide expansion with 66 or 149 repeats [74, 75]. In the 66-repeat model, the mice show evident expression of intranuclear sense RNA foci and DPRs in the central nervous system (CNS), accompanied by motor dysfunction and anxiety-like behaviors, as well as cortical neuronal deficiency [75]. Strikingly, nuclear and cytoplasmic phosphorylated TDP-43 inclusions have been observed in the cortex and hippocampus of (G4C2)66 mice [75]. The 149-repeat mice show similar phenotypes, while antisense RNA foci, in amounts from 10% to 20%, have been found in the hippocampus, cortex, and cerebellum. In addition, the antisense DPRs poly-PA and poly-PR have been detected in the cortex of 149-repeat mice at 3 months of age [74]. Mis-localization of RanGAP1 and aggregation of stress granule-associated proteins have also been reported in the cortex and hippocampus of 149-repeat mice [74], suggesting that overexpression of HRE is enough to model the neuropathological changes of c9ALS/FTD.
To identify the pathological effects of HRE at lower expression levels, several groups have developed bacterial artificial chromosome (BAC) transgenic mouse models [29, 76,77,78]. Despite RNA foci and a subset of DPRs in mice containing 500 or 100–1000 repeats of the G4C2 sequence, two BAC models show no behavioral deficiency or neurodegeneration [76, 77]. However, two other transgenic mice show a clear phenotype [29, 78]. One BAC mouse embedded with a patient-derived C9ORF72 gene harbor either 110 or 450 repeats of G4C2, and the 450-repeat mice display cognitive impairment but not motor deficits, accompanied by size- and dose-dependent expression of RNA foci and DPRs in the CNS [29]. Other transgenic mouse models that have the full-length human C9ORF72 gene with ~30 and ~500 repeats show obvious gait abnormalities, anxiety-like behavior, and decreased survival, as well as widespread neurodegeneration and TDP-43 pathology [78]. Notably, the antisense RNA foci preferentially accumulate in the c9ALS/FTD-vulnerable cell populations [11, 79, 80]. As both of the latter models have higher expression of human C9ORF72 mRNA levels than those in the former models [29, 76,77,78], the expression levels of the human C9ORF72 gene in mice and the antisense RNA foci and related DPRs may contribute to the phenotypes.
Disease Models with Dipeptide Repeat Proteins
The G4C2 expansion can be translated into five DPRs in a repeat-associated non -ATG-initiated manner (poly-GA, poly-GP, poly-GR, poly-PA, and poly-PR) [7, 9,10,11]. Among these, poly-GP and poly-GA display higher expression in the c9ALS/FTD brain than the other three DPRs [53]. As poly-GP and poly-PA have been reported to have no cytotoxicity [22, 23, 81], most studies focus on the roles of poly-GA, poly-GR, and poly-PR in neurodegeneration.
Role of Poly-GA in Cytotoxicity
Cell-Culture Models of Poly-GA
Poly-GA forms cellular inclusions and co-localizes with ubiquitin and p62 in GFP-GA50-transfected cultured cells [82]. Poly-GA overexpression induces cytotoxicity, including an increase in caspase-3-positive cells and release of lactate dehydrogenase, which is also found in cultured primary cortical neurons overexpressing poly-GA [82]. In primary hippocampal neurons, a long repeat length of poly-GA (149 repeats) forms p62-positive inclusions and induces dendrite loss [83]. In addition, GA50 and GA100 cause slight neuronal toxicity in Neuro-2a [81] and NSC-34 cells [84]. However, some studies have reported that 30-repeat lengths of GA have no significant toxicity in NSC-34 and HEK293 cells [24], and that both GA and PA are not neurotoxic even at a length of 200 repeats [23]. To date, it is unclear whether the toxicity of poly-GA depends on the expression levels or cell types.
Drosophila Models with Poly-GA
To determine the role of poly-GA in neurodegeneration, several groups have established poly-GA Drosophila models with different repeat lengths [22, 23, 81]. The expression of GA50 and PA50 in Drosophila does not induce neurotoxicity, which is consistent with the previous finding in primary hippocampal neurons [23]. Moreover, GA100 and PA100 have no effects on the egg-to-adult viability of Drosophila at different temperatures, although GA100 causes a late-onset decrease in survival [22], suggesting a mild toxicity of poly-GA. In addition, overexpression of GA80 does not induce degeneration in Drosophila eyes or wing margins [85].
Zebrafish and Chicken Models with Poly-GA
Although poly-GA in Drosophila does not show cellular toxicity, zebrafishes expressing GA80 show a strong pericardial edema phenotype and dramatically decreased circulation, accompanied by an accumulation of red blood cells, and these phenotypes can be rescued by interfering with the expression of GA80, indicating a toxic property of poly-GA in zebrafish [86]. In addition, poly-GA shows the highest toxicity to neurons in the spinal cord of transgenic chickens as compared to other DPRs [87]. Poly-GA sequesters other DPRs to its aggregates and overexpression of poly-PA inhibits poly-GA aggregation [87], suggesting a role of GA in aggregate formation and the influence of other DPRs on GA aggregation.
Mouse Models with Poly-GA
Given the limitations of cultured cells and Drosophila models, several groups have constructed poly-GA transgenic mouse models [88,89,90]. In poly-GA transgenic mice, in which the expression of poly-GA is controlled by the Thy1 promoter, poly-GA is mainly distributed in the spinal cord and brainstem [88]. The mice show co-aggregation of poly-GA with p62, Rad23b, and Mlf2; this also occurs in c9ALS/FTD patients [88]. The animals show some behavioral changes, including motor imbalance and hypoactivity, but no defects in muscle strength or spatial memory [88]. Moreover, the mice have no significant motor neuron deficiency in the spinal cord, although microglial activation is present there [88]. With intracerebroventricular injection of adeno-associated virus (AAV)-packaged GFP-GA50, the mice present motor deficits, brain atrophy, neurodegeneration, and neuroinflammation at post-natal day 0 [89]. Poly-GA-induced motor dysfunction has been confirmed using a similar method [90]. Thus, overexpressed poly-GA has neurotoxic effects and induces motor defects in vertebrates, although its effects in cultured cells or Drosophila remain controversial.
Roles of Poly-GR and Poly-PR in Cytotoxicity
Cellular Models with Poly-GR and Poly-PR
Within DPRs, poly-GR and poly-PR show strong toxicity in both cellular and animal models, in which several methods have been applied to avoid the toxic effects caused by G4C2 RNA foci. In vitro analyses show that synthetic GR20 or PR20 forms nucleolar inclusions and is significantly cytotoxic in U2OS cells and primary astrocytes [21]. In primary cortical neurons, overexpression of PR50 has high neurotoxicity, while poly-GR-induced cell death is repeat-length dependent [23]. Moreover, the cellular toxicity of arginine-rich poly-GR and poly-PR has been reported in multiple cell lines, including Neuro-2a [81], NSC-34 [24, 84], SH-SY5Y [91], and primary cortical neurons [92]. In addition, overexpression of PR100 [93] and GR80 [94] in induced pluripotent stem cell (iPSC)-derived neurons induces significant neuron deficiency and a DNA damage response, further indicating the high toxicity of arginine-rich poly-GR and poly-PR.
Drosophila Models with Poly-GR and Poly-PR
To address whether DPR-induced neurotoxicity depends on repeat RNA, Mizielinska and colleagues have constructed Drosophila models in which they use two strategies: (1) a 6 base-pair interruption that contains a stop codon in both the sense and antisense directions is inserted in every G4C2 repeat, which construct produces “RNA-only” repeats without DPR expression; and (2) using alternative codons that encode the same amino-acids but disrupt the G4C2 repeat, which construct expresses “DPR-only” without G4C2 repeats [22]. In Drosophila, GR100 and PR100 exhibit strong toxicity, leading to severe neurodegeneration and reduced survival, while GA100 causes late-onset disease [22]. However, “RNA-only” repeats do not induce neurodegeneration and disease phenotypes, suggesting that DPRs, especially poly-GR and poly-PR, are major factors in C9ORF72-induced pathogenesis [22]. Expression of PR50 in motor neurons induces developmental failure in Drosophila, despite a normal body morphology [23]. In addition to the neuronal toxicity of arginine-rich DPRs, GR80 exhibits non-neuronal cellular toxicity in Drosophila, such as wing margin defects [85], suggesting that PR and GR are highly toxic to both neuronal and non-neuronal cells in Drosophila.
Mouse Models with Poly-GR and Poly-PR
Several C9ORF72 BAC transgenic mice have been established to identify the role of the C9ORF72 gene in disease [29, 76,77,78]. Nuclear RNA foci and DPRs have been found in the brain of BAC transgenic mice that have no motor neuron degeneration, although one BAC mouse with an FVB/NJ background shows decreased survival and motor deficits [29, 76,77,78]. Thus, transgenic mice with long hexanucleotide repeats in the C9ORF72 gene do not present disease phenotypes, or have only mild phenotypes. Due to the high toxicity of poly-GR and poly-PR in cultured cells, iPSC-derived neurons, and Drosophila models, arginine-rich DPR mouse models have been created [95,96,97]. With intracerebroventricular injections of AAV1 that expresses GFP-GR100, the GFP-GR100 is mainly expressed in the CNS, with a cytoplasmic and diffuse distribution, while there is little expression in the spinal cord [95]. The GR100 mice display progressive motor deficits and memory loss accompanied by age-dependent cortical and hippocampal neurodegeneration. In addition, glial activation occurs at 1.5 months of age [95]. In another mouse model, the spatial and temporal expression of poly-GR is controlled by Tet expression systems, and the expression of GR80 is mainly distributed in the frontal cortex relative to other cortical regions, with diffuse distribution in the cytoplasm and nucleus of 95% of neurons [96]. In addition, the transgenic mice display age-dependent social behavioral deficits, impaired synaptic transmission, cortical neuronal loss, and microglial activation, but no changes in body weight, locomotor activity, and working memory [96]. Thus studies suggest that a long repeat length of poly-GR has a diffuse cytoplasmic distribution and is able to induce severe neurodegeneration and related behavioral deficits in vivo.
The poly-PR that shows the highest neurotoxicity in cellular and Drosophila models has toxic effects in poly-PR AAV-infected mice and transgenic mice [55, 97]. AAV-infected GFP-PR50 mice show behavioral deficiencies and neurodegeneration at an early stage [55]. Moreover, the overexpression of poly-PR in neurons is highly toxic; up to 60% of GFP-PR50-expressing mice die by 4 weeks of age [55]. Strikingly, in this AAV-mediated poly-PR expressing mouse, besides a nucleolar distribution, poly-PR mainly exhibits heterochromatic localization, which elicits aberrant post-translational modifications of histone H3 [55]. In the poly-PR transgenic mice with intermediate repeat lengths of poly-PR, the neuronal expression of GFP-PR28 driven by Cre recombinase under control of the Thy1 promoter is highly toxic [97]. Homozygous transgenic mice develop body weight loss and premature death, similar to GFP-PR50 AAV mice [97]. Heterozygous mice exhibit age-dependent motor dysfunction, decreased survival time, motor-related neuronal deficiency, and neuroinflammation [97]. Unlike the heterochromatic localization of GFP-PR50 in AAV mice [55], GFP-PR28 is mainly distributed in the nucleolus of neurons, as well as a diffuse cytoplasmic distribution in lumbar spinal motor neurons [97]. It is still unknown whether the difference of repeat length in poly-PR between these two models leads to the difference in poly-PR distribution. The animal models of C9ORF72 with either HRE or dipeptide repeat proteins are summarized in Table 2.
In addition to the difference in cellular localization between poly-GA and arginine-rich DPRs [24], poly-GR and -PR are more neurotoxic than poly-GA in cultured cells [24] and Drosophila [22]. Consistent with this, poly-GA mainly influences the cytoplasmic ubiquitin-proteasome system [98], while poly-GR and -PR have effects on nuclear processes. Although all these DPR mice display serious behavioral deficits and neurodegeneration [55, 95,96,97], they do not develop TDP-43 pathology that is a general neuropathological feature of c9ALS/FTD [56,57,58]. These data also suggest that the pathogenesis in patients may be complicated. Synergic effects, such as cooperativity between gain- and loss-of-function mechanisms, may be essential for the induction of pathological events.
Pathological Mechanisms
Since the discovery of the non-ATG-initiated translation of C9ORF72 repeat expansions, a variety of pathological mechanisms linked to DPRs have been identified, ranging from DNA processes to RNA processing to protein translation (Fig. 2).

Potential mechanisms linked to DPR overexpression. DPR overexpression causes neuronal deficiency through mechanisms that range from DNA processes to RNA processing to protein translation. DPRs cause a DNA damage response and poly-PR induces abnormal histone methylation and dysfunction of heterochromatin. Defects in RNA processing such as nonsense-mediated RNA decay, RNA splicing, and ribosomal RNA processing are linked to arginine-rich DPRs. Moreover, these DPRs contribute to dysfunction of protein homeostasis through nucleocytoplasmic transport, the unfolding protein response (ER-stress), and translation. The diagram in the lower right corner summarizes the biological processes related to poly-PR-binding proteins, which mainly include the nucleolus, RNA splicing, the nuclear pore complex, and the ribosome. rDNA, ribosomal DNA; rRNA, ribosomal RNA.
Shedding Light on DNA Processes
DNA Damage Response (DDR)
Previous studies have demonstrated that an impaired DDR is essential for neuronal deficiency in neurodegenerative diseases [99, 100]. Individuals with mutations in XRCC1 that encodes a scaffold protein that is involved in DNA single-strand break repair, present ocular motor apraxia, axon neuropathy, and progressive cerebellar ataxia [101]. Mutations in another ALS/FTD-related gene FUS (Fused-in-Sarcoma) lead to a DDR and DNA repair dysfunction [102,103,104,105,106]. Impairment of the DDR can also be induced by mutations in TDP-43 [107, 108], ATM [109], APTX [110], and other genes, suggesting a role of the DDR in neurodegeneration.
An essential role of the DDR in the pathogenesis of c9ALS/FTD has been documented in several studies that demonstrated genomic instability as a key event in C9ORF72-linked neurodegeneration. In iPSC-derived C9ORF72 motor neurons, there is increased expression of γH2AX, a marker of DNA double-strand breaks, and increased tail length in the comet assay, suggesting the presence of DNA damage in c9ALS/FTD [94]. Moreover, the expression of γH2AX is also increased in the spinal motor neurons of C9ORF72 patients [90, 93]. In primary cortical neurons and the SH-SY5Y cell line, overexpression of poly-GR and poly-PR induces an accumulation of γH2AX foci and an increase of phosphorylated ATM, the main kinase for DNA repair [93]. Moreover, in poly-GA and repeat RNA overexpression cellular models, the levels of DNA–RNA hybrids (R-loops), a three-stranded nucleic acid structure produced during the transcription of repeat sequences, are increased, which can lead to genome instability [90]. Furthermore, the ATM signaling pathway is impaired in poly-GA transgenic mice, suggesting that defects in the DNA repair machinery are associated with DPR and RNA repeat-induced DNA damage [90]. DNA damage is increased in iPSC-derived C9ORF72 motor neurons [94]. Overexpression of poly-GR but not poly-GA in control iPSC-derived motor neurons induces DNA damage [94], further suggesting a role of DPRs in DNA damage.
Heterochromatin and Histone Methylation
Poly-PR is localized to heterochromatin in the cortex of transgenic mice as well as in c9ALS/FTD patients where it causes abnormal histone methylation [55]. In addition, poly-PR reduces the expression levels of HP1α and disrupts the phase separation of HP1α, leading to lamin invaginations and double-stranded RNA accumulation [55]. Meanwhile, repetitive elements that make up a large portion of heterochromatin are broadly upregulated in the brains of c9ALS/FTD patients and mice overexpressing poly-PR [55, 111].The upregulation of abnormal repetitive elements and accumulation of double-stranded RNA induce neurodegeneration. Thus, the abnormality of histone methylation and the dysfunction of heterochromatin and DNA components induced by poly-PR may contribute to the neurodegeneration in c9ALS/FTD.
RNA Processing
Nonsense-Mediated RNA Decay (NMD)
NMD, a cellular RNA degradation system that is activated in response to stress, rapidly degrades mRNA containing premature termination codons to prevent the translation of defective proteins [112, 113]. UPF1–3 are master factors for NMD activation in which UPFs interact with each other, the ribosome, and multiple mRNA decay factors [112, 113]. NMD can be inhibited, which is indicated by an overlap of upregulated NMD substrate genes in c9ALS/FTD-derived iPSCs and UPF3B-/-lymphocytes [91]. Numerous NMD substrate genes accumulate in PR36 Drosophila [91]. In addition, there are significant overlaps in the accumulated NMD genes in flies overexpressing PR36 and those deficient in UPF1 or UPF2 [91]. Furthermore, the NMD substrate genes Sin3A, Gadd45, Xrp1, and Arc1 do not accumulate in flies expressing α-synuclein, Htt-128Q, or human FUS, although these genes do accumulate in flies overexpressing GR36 or PR36 or with UPF1 knockdown [91]. In contrast, overexpression of UPF1 significantly inhibits the neurotoxicity induced by GR36 or PR36.
Although the primary cause of NMD by DPRs remains unknown, a reactivation of NMD significantly ameliorates the neurotoxicity in flies expressing DPRs, indicating that impairment of NMD contributes to the pathology of C9ORF72.
RNA Splicing
Pre-mRNA splicing and the biogenesis of rRNA are significantly impaired in cultured human astrocytes expressing poly-PR/GR [21]. Notably, FUS and TDP-43, two other ALS-related gene products, have been shown to cause a pre-mRNA splicing dysfunction and reduce downstream gene expression [114,115,116,117]. Another study using unbiased quantitative mass spectrometry demonstrated that poly-GR/PR binds to U2 snRNPs (small nuclear ribonucleoproteins) and inhibits spliceosome assembly [118]. Moreover, bioinformatics analysis showed that U2-dependent exons are mis-spliced in the cortex and cerebellum of C9ORF72 patients [118]. Previous results have revealed that arginine-rich poly-PR and poly-GR, but not poly-GA, bind to proteins that contain low complexity domains [81, 119]. Consistently, a number of binding proteins with low complexity domains, including several U2 snRNPs, have also been detected [118]. These data suggest that U2 snRNPs sequestered by arginine-rich DPRs contribute to the blocked alternative splicing in C9ORF72 patients.
Protein Homeostasis
Proteinopathies such as ALS mainly exhibit protein inclusions with impaired quality control systems in cells [13, 14, 120]. Poly-GR and poly-PR interact with proteins that contain low complexity domains and disturb the phase separation of membrane-less organelles, such as the nucleolus, the nuclear pore complex, and stress granules [81, 119, 121, 122]. The function of poly-GR and poly-PR in the nuclear pore complex and stress granules has been systematically described in other reviews [123, 124]. Here, we mainly focus on the role of poly-GR and poly-PR in the protein quality control system, including the unfolded protein response and translation.
Unfolded Protein Response (UPR)
Using unbiased CRISPR-Cas9 screens, TMX2, an endoplasmic reticulum (ER)-resident transmembrane thioredoxin protein, has been identified as a major modifier of neurotoxicity in primary cortical neurons infected with poly-PR [92]. Moreover, RNA-sequencing analysis has demonstrated that the genes in the ER-stress/UPR pathway are significantly upregulated, including Atf4, Bbc3, and Chac1. Knockdown of TMX2 ameliorates the neurotoxicity caused by poly-PR overexpression in primary cortical neurons and in motor neurons derived from C9ORF72 patients [92]. Notably, the ER-stress-related genes Chac1 and Atf5 are upregulated in the cerebellum of poly-PR transgenic mice at 2 months of age, before motor dysfunction occurs, suggesting that ER-stress is an early event in the pathogenesis of poly-PR [97, 125]. In addition, poly-GA overexpression induces pathological inclusions, neurotoxicity, and ER-stress [82]. Pharmacological inhibitors of ER-stress decrease the expression of ER-stress markers and alleviate the neurotoxicity caused by poly-GA [62]. Besides c9ALS/FTD, SOD1-mutated ALS and other neurodegenerative diseases also show the presence of ER-stress in the CNS of patients and animal models [126,127,128]. Interestingly, using techniques that combine translational ribosome affinity purification and high-throughput RNA sequencing, a cascade of cell type-specific dysregulated processes in SOD1-mutated mice has been identified, showing that the UPR starts within neurons, followed by metabolic and inflammatory gene changes in astrocytes and membrane protein gene alteration in oligodendrocytes [126]. Thus, ER-stress is an early reaction occurring specifically in motor neurons in ALS, and maybe a potential target in ALS therapy.
One interesting but still unanswered question is how poly-PR induces the UPR, as arginine-rich poly-PR is localized in the nucleolus, not the ER. Recently, the nucleolus has been identified as a phase-separated protein control compartment [129]. Under stress, nucleoplasmic proteins are transported to the nucleolus, where Hsp70 mediates refolding, preventing the irreversible aggregation of misfolded proteins [129]. In cells expressing poly-PR, disaggregation of misfolded proteins is inhibited and mobile fractions of liquid-separated proteins are significantly reduced, suggesting an inhibition of nucleolar quality control [129]. Thus, misfolded protein aggregation in the nucleolus, which is caused by poly-PR overexpression, may contribute to the misfolded protein response in c9ALS/FTD. However, it remains unknown whether the nucleolus transmits a stress signal to the ER, thereby leading to ER-stress.
Translation
It is well documented that arginine-rich poly-GR and poly-PR interact with ribosomal proteins, and proteins involved in translation [81, 94, 130, 131]. An inhibition of global translation is consistently found in cultured cells expressing poly-PR [132], primary rat cortical neurons [130], adult Drosophila [131], and human iPSC-derived motor neurons [131]. Overexpression of the translation initiation factor eIF1A rescues the translational defects and neurotoxicity caused by poly-PR [131]. In addition, overexpression of poly-GR100 in mice causes significant neurodegeneration with a dysregulation of genes involved in the ribosomal pathway [95]. The impaired canonical translation occurs in neurons of the GR-expressing mouse model; besides, the non-canonical translation (repeat-associated non-ATG-initiated translation, RAN translation) is also found [95], which argues against previous studies suggesting that DPR-induced ER-stress contributes to the selective activation of RAN translation in vitro [133,134,135]. One reason for the difference may be that the expression of DPR-induced ER-stress thereby activates RAN translation at an early stage, while RAN translation is inhibited under chronic stress.
Therapeutic Advances
Due to the high frequency of C9ORF72 mutation and the urgent requirement for clinical treatment in ALS and FTD, a wide variety of efforts have been put into the identification of promising therapeutic approaches, from DNA to RNA to DPR protein processes.
DNA Processes
DNA methylation plays a pivotal role in regulating the expression of downstream genes. Hypermethylation has been found in the promoter of the C9ORF72 gene, which contains G4C2 repeat expansions, leading to reduced C9orf72 mRNA levels [136]. Neurons from carriers of hypermethylated C9ORF72 repeat expansions display reduced RNA foci and DPR aggregations, suggesting a protective role of hypermethylation in C9ORF72-related pathology [136]. Similarly, genetic manipulation using CRISPR-Cas9 technology that targets the promoter region of the C9ORF72 gene decreases the levels of C9ORF72 and DPR proteins, and ameliorates neurotoxicity in iPSC-derived neurons [137]. Despite the promising effects, further efforts are needed to avoid the adverse effects of downregulation of C9ORF72 protein that is essential for immune function [16] and protein trafficking [28].
RNA Processing
Using unbiased large-scale screens, a number of possible remedies for c9ALS/FTD have focused on transcriptional regulation, targeting the transcriptional regulator PAF1 [138], transcriptional elongation factor SPT4 [139], and AFF2/FMR2 [140]. In addition, antisense oligonucleotides (ASOs) that are single-stranded show clear alleviation of nuclear RNA foci and neurotoxicity [19, 141, 142]. Given the essential role of normal C9ORF72 protein, optimized ASOs exhibiting no C9ORF72 RNA reduction have been designed and are successful in reducing the number of sense RNA foci and the expression of DPRs in transgenic mouse models [29], indicating a hopeful outlook for ASOs in the treatment of ALS and FTD. In addition, RNA-targeting Cas9 is also effective in eliminating the G4C2 repeat RNA foci [143].
Protein Homeostasis
As a result of the high toxicity of DPRs, several strategies have been designed to reduce their levels. RPS25 [144], a small ribosomal protein subunit, and DDX3X [145], an RNA helicase, suppress the translation of DPRs and improve the survival of patient-derived induced motor neurons. Moreover, specific antibodies targeting poly-GA significantly reduce GA protein levels, ameliorate neuronal deficiency, and improve motor dysfunction in transgenic mice [146, 147]. Therefore, selective targeting of DPR expression may also be an alternative therapeutic approach.
Conclusions and Perspectives
Despite the broad achievements in exploring the role of DPRs in disease, several key questions remain unanswered. The DPRs form neuronal cytoplasmic inclusions that are widespread in c9ALS/FTD patients, while overexpression of DPRs in vivo, especially arginine-rich poly-PR, only show intranuclear inclusions in selective neurons. It remains unknown whether the repeat length of poly-PR, or the localization of other DPRs that may interact with poly-PR, contributes to the cellular distribution. Similarly, the distribution of TDP-43 is inconsistent with the pathological features and a priority is to identify which factors are essential for TDP-43 inclusions, RNA foci, or aggregated DPRs. It will also be interesting to determine whether DPR inclusions in muscle contribute to clinical pathology [148, 149].
References
Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 2004, 27: 723–749.
Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet 2015, 386: 1672–1682.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72: 245–256.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72: 257–268.
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 2012, 11: 54–65.
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77: 639–646.
Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 2013, 126: 829–844.
Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol 2013, 126: 845–857.
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 2013, 126: 881–893.
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339: 1335–1338.
Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 2013, 110: E4968–4977.
Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol 2013, 126: 859–879.
Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J 2016, 35: 1276–1297.
Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 2016, 35: 1656–1676.
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 2016, 8: 347ra393.
O’Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351: 1324–1329.
Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CA, et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol 2016, 132: 145–147.
Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 2013, 5: 1178-1186.
Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80: 415–428.
Swinnen B, Bento-Abreu A, Gendron TF, Boeynaems S, Bogaert E, Nuyts R, et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol 2018, 135: 427–443.
Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345: 1139–1145.
Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345: 1192–1194.
Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84: 1213–1225.
Tao Z, Wang H, Xia Q, Li K, Li K, Jiang X, et al. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet 2015, 24: 2426–2441.
Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun 2016, 4: 51.
Ugolino J, Ji YJ, Conchina K, Chu J, Nirujogi RS, Pandey A, et al. Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet 2016, 12: e1006443.
Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv 2016, 2: e1601167.
Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 2018, 24: 313–325.
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 2016, 90: 535–550.
van Blitterswijk M, DeJesus-Hernandez M, Rademakers R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol 2012, 25: 689–700.
Rademakers R, van Blitterswijk M. Motor neuron disease in 2012: Novel causal genes and disease modifiers. Nat Rev Neurol 2013, 9: 63–64.
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012, 11: 323–330.
Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2017, 88: 540–549.
Liu X, He J, Gao FB, Gitler AD, Fan D. The epidemiology and genetics of Amyotrophic lateral sclerosis in China. Brain Res 2018, 1693: 121–126.
Liu Q, Liu F, Cui B, Lu CX, Guo XN, Wang RR, et al. Mutation spectrum of Chinese patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2016, 87: 1272–1274.
Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, et al. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry 2011, 82: 196–203.
Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012, 135: 765–783.
Chio A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 2012, 135: 784–793.
Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 2012, 135: 751–764.
Hsiung GY, DeJesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 2012, 135: 709–722.
Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain 2012, 135: 736–750.
Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 2012, 135: 723–735.
Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 2012, 135: 693–708.
Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus-Hernandez M, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 2012, 135: 794–806.
Umoh ME, Fournier C, Li Y, Polak M, Shaw L, Landers JE, et al. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology 2016, 87: 1024–1030.
Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, et al. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 2010, 53: 1070–1076.
Spina S, Farlow MR, Unverzagt FW, Kareken DA, Murrell JR, Fraser G, et al. The tauopathy associated with mutation +3 in intron 10 of Tau: characterization of the MSTD family. Brain 2008, 131: 72–89.
Beck J, Rohrer JD, Campbell T, Isaacs A, Morrison KE, Goodall EF, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 2008, 131: 706–720.
Whitwell JL, Jack CR, Jr., Baker M, Rademakers R, Adamson J, Boeve BF, et al. Voxel-based morphometry in frontotemporal lobar degeneration with ubiquitin-positive inclusions with and without progranulin mutations. Arch Neurol 2007, 64: 371–376.
Hodges J. Familial frontotemporal dementia and amyotrophic lateral sclerosis associated with the C9ORF72 hexanucleotide repeat. Brain 2012, 135: 652–655.
Corcia P, Vourc’h P, Guennoc AM, Del Mar Amador M, Blasco H, Andres C, et al. Pure cerebellar ataxia linked to large C9orf72 repeat expansion. Amyotroph Lateral Scler Frontotemporal Degener 2016, 17: 301–303.
Goldman JS, Quinzii C, Dunning-Broadbent J, Waters C, Mitsumoto H, Brannagan TH, 3rd, et al. Multiple system atrophy and amyotrophic lateral sclerosis in a family with hexanucleotide repeat expansions in C9orf72. JAMA Neurol 2014, 71: 771–774.
Mackenzie IR, Frick P, Grasser FA, Gendron TF, Petrucelli L, Cashman NR, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol 2015, 130: 845–861.
Schludi MH, May S, Grasser FA, Rentzsch K, Kremmer E, Kupper C, et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol 2015, 130: 537–555.
Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 2019, 363: eaav2606.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314: 130–133.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319: 1668–1672.
Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol 2009, 117: 137–149.
Burrell JR, Halliday GM, Kril JJ, Ittner LM, Gotz J, Kiernan MC, et al. The frontotemporal dementia-motor neuron disease continuum. Lancet 2016, 388: 919–931.
Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol 2011, 122: 673–690.
Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, et al. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 2012, 123: 409–417.
Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J, et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol 2013, 126: 401–409.
Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol 2011, 122: 691–702.
Troakes C, Maekawa S, Wijesekera L, Rogelj B, Siklos L, Bell C, et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology 2012, 32: 505–514.
Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 2015, 18: 1226–1229.
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525: 129–133.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525: 56–61.
Shi KY, Mori E, Nizami ZF, Lin Y, Kato M, Xiang S, et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc Natl Acad Sci U S A 2017, 114: E1111–E1117.
Gorlich D, Pante N, Kutay U, Aebi U, Bischoff FR. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J 1996, 15: 5584–5594.
Ritterhoff T, Das H, Hofhaus G, Schroder RR, Flotho A, Melchior F. The RanBP2/RanGAP1*SUMO1/Ubc9 SUMO E3 ligase is a disassembly machine for Crm1-dependent nuclear export complexes. Nat Commun 2016, 7: 11482.
Burguete AS, Almeida S, Gao FB, Kalb R, Akins MR, Bonini NM. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife 2015, 4: e08881.
Celona B, Dollen JV, Vatsavayai SC, Kashima R, Johnson JR, Tang AA, et al. Suppression of C9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. Elife 2017, 6: e19032.
Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A 2013, 110: 7778–7783.
Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener 2019, 14: 9.
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348: 1151–1154.
O’Rourke JG, Bogdanik L, Muhammad A, Gendron TF, Kim KJ, Austin A, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron 2015, 88: 892–901.
Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 2015, 88: 902–909.
Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, et al. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 2016, 90: 521–534.
Cooper-Knock J, Higginbottom A, Stopford MJ, Highley JR, Ince PG, Wharton SB, et al. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol 2015, 130: 63–75.
Aladesuyi Arogundade O, Stauffer JE, Saberi S, Diaz-Garcia S, Malik S, Basilim H, et al. Antisense RNA foci are associated with nucleoli and TDP-43 mislocalization in C9orf72-ALS/FTD: a quantitative study. Acta Neuropathol 2019, 137: 527–530.
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 2016, 167: 774–788 e717.
Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 2014, 128: 505–524.
May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 2014, 128: 485–503.
Suzuki H, Shibagaki Y, Hattori S, Matsuoka M. The proline-arginine repeat protein linked to C9-ALS/FTD causes neuronal toxicity by inhibiting the DEAD-box RNA helicase-mediated ribosome biogenesis. Cell Death Dis 2018, 9: 975.
Yang D, Abdallah A, Li Z, Lu Y, Almeida S, Gao FB. FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol 2015, 130: 525–535.
Ohki Y, Wenninger-Weinzierl A, Hruscha A, Asakawa K, Kawakami K, Haass C, et al. Glycine-alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration. Mol Neurodegener 2017, 12: 6.
Lee YB, Baskaran P, Gomez-Deza J, Chen HJ, Nishimura AL, Smith BN, et al. C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum Mol Genet 2017, 26: 4765–4777.
Schludi MH, Becker L, Garrett L, Gendron TF, Zhou Q, Schreiber F, et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol 2017, 134: 241-254.
Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci 2016, 19: 668–677.
Walker C, Herranz-Martin S, Karyka E, Liao C, Lewis K, Elsayed W, et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat Neurosci 2017, 20: 1225–1235.
Xu W, Bao P, Jiang X, Wang H, Qin M, Wang R, et al. Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. Brain 2019, 142: 1349–1364.
Kramer NJ, Haney MS, Morgens DW, Jovicic A, Couthouis J, Li A, et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat Genet 2018, 50: 603–612.
Farg MA, Konopka A, Soo KY, Ito D, Atkin JD. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet 2017, 26: 2882–2896.
Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, et al. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 2016, 92: 383–391.
Zhang YJ, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen-West K, et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med 2018, 24: 1136–1142.
Choi SY, Lopez-Gonzalez R, Krishnan G, Phillips HL, Li AN, Seeley WW, et al. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat Neurosci 2019, 22: 851–862.
Hao Z, Liu L, Tao Z, Wang R, Ren H, Sun H, et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat Commun 2019, 10: 2906.
Guo Q, Lehmer C, Martinez-Sanchez A, Rudack T, Beck F, Hartmann H, et al. In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 2018, 172: 696–705 e612.
El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005, 434: 108–113.
Enokido Y, Tamura T, Ito H, Arumughan A, Komuro A, Shiwaku H, et al. Mutant huntingtin impairs Ku70-mediated DNA repair. J Cell Biol 2010, 189: 425–443.
Hoch NC, Hanzlikova H, Rulten SL, Tetreault M, Komulainen E, Ju L, et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 2017, 541: 87–91.
Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, et al. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 2013, 16: 1383–1391.
Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem 2013, 288: 24731–24741.
Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DA, Gomez-Herreros F, et al. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res 2014, 42: 307–314.
Naumann M, Pal A, Goswami A, Lojewski X, Japtok J, Vehlow A, et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun 2018, 9: 335.
Wang H, Guo W, Mitra J, Hegde PM, Vandoorne T, Eckelmann BJ, et al. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat Commun 2018, 9: 3683.
Mitra J, Guerrero EN, Hegde PM, Liachko NF, Wang H, Vasquez V, et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A 2019, 116: 4696–4705.
Hill SJ, Mordes DA, Cameron LA, Neuberg DS, Landini S, Eggan K, et al. Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc Natl Acad Sci U S A 2016, 113: E7701–E7709.
Dar I, Biton S, Shiloh Y, Barzilai A. Analysis of the ataxia telangiectasia mutated-mediated DNA damage response in murine cerebellar neurons. J Neurosci 2006, 26: 7767–7774.
Hirano M, Yamamoto A, Mori T, Lan L, Iwamoto TA, Aoki M, et al. DNA single-strand break repair is impaired in aprataxin-related ataxia. Ann Neurol 2007, 61: 162–174.
Prudencio M, Gonzales PK, Cook CN, Gendron TF, Daughrity LM, Song Y, et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum Mol Genet 2017, 26: 3421–3431.
He F, Jacobson A. Nonsense-mediated mRNA decay: Degradation of defective transcripts is only part of the story. Annu Rev Genet 2015, 49: 339–366.
Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 2015, 16: 665–677.
Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 2011, 14: 459–468.
Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 2012, 15: 1488–1497.
Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A 2013, 110: E736–745.
Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun 2015, 6: 6171.
Yin S, Lopez-Gonzalez R, Kunz RC, Gangopadhyay J, Borufka C, Gygi SP, et al. Evidence that C9ORF72 dipeptide repeat proteins associate with U2 snRNP to cause mis-splicing in ALS/FTD patients. Cell Rep 2017, 19: 2244–2256.
Lin Y, Mori E, Kato M, Xiang S, Wu L, Kwon I, et al. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 2016, 167: 789–802 e712.
Wu D, Hao Z, Ren H, Wang G. Loss of VAPB regulates autophagy in a Beclin 1-dependent manner. Neurosci Bull 2018, 34: 1037–1046.
Boeynaems S, Bogaert E, Kovacs D, Konijnenberg A, Timmerman E, Volkov A, et al. Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol Cell 2017, 65: 1044–1055 e1045.
White MR, Mitrea DM, Zhang P, Stanley CB, Cassidy DE, Nourse A, et al. C9orf72 Poly(PR) dipeptide repeats disturb biomolecular phase separation and disrupt nucleolar function. Mol Cell 2019, 74: 713–728 e716.
Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci 2016, 17: 383–395.
Gao FB, Almeida S, Lopez-Gonzalez R. Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J 2017, 36: 2931–2950.
Wang R, Xu X, Hao Z, Zhang S, Wu D, Sun H, et al. Poly-PR in C9ORF72-related amyotrophic lateral sclerosis/frontotemporal dementia causes neurotoxicity by clathrin-dependent endocytosis. Neurosci Bull 2019, 35: 889–900.
Sun S, Sun Y, Ling SC, Ferraiuolo L, McAlonis-Downes M, Zou Y, et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci U S A 2015, 112: E6993–7002.
Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 2014, 15: 233–249.
Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol 2006, 18: 444–452.
Frottin F, Schueder F, Tiwary S, Gupta R, Korner R, Schlichthaerle T, et al. The nucleolus functions as a phase-separated protein quality control compartment. Science 2019, 365: 342–347.
Hartmann H, Hornburg D, Czuppa M, Bader J, Michaelsen M, Farny D, et al. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci Alliance 2018, 1: e201800070.
Moens TG, Niccoli T, Wilson KM, Atilano ML, Birsa N, Gittings LM, et al. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol 2019, 137: 487–500.
Kanekura K, Yagi T, Cammack AJ, Mahadevan J, Kuroda M, Harms MB, et al. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet 2016, 25: 1803–1813.
Cheng W, Wang S, Mestre AA, Fu C, Makarem A, Xian F, et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2alpha phosphorylation. Nat Commun 2018, 9: 51.
Tabet R, Schaeffer L, Freyermuth F, Jambeau M, Workman M, Lee CZ, et al. CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat Commun 2018, 9: 152.
Green KM, Glineburg MR, Kearse MG, Flores BN, Linsalata AE, Fedak SJ, et al. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat Commun 2017, 8: 2005.
Liu EY, Russ J, Wu K, Neal D, Suh E, McNally AG, et al. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol 2014, 128: 525–541.
Krishnan G, Zhang Y, Gu Y, Kankel MW, Gao FB, Almeida S. CRISPR deletion of the C9ORF72 promoter in ALS/FTD patient motor neurons abolishes production of dipeptide repeat proteins and rescues neurodegeneration. Acta Neuropathol 2020, 140: 81–87.
Goodman LD, Prudencio M, Kramer NJ, Martinez-Ramirez LF, Srinivasan AR, Lan M, et al. Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat Neurosci 2019, 22: 863–874.
Kramer NJ, Carlomagno Y, Zhang YJ, Almeida S, Cook CN, Gendron TF, et al. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science 2016, 353: 708–712.
Yuva-Aydemir Y, Almeida S, Krishnan G, Gendron TF, Gao FB. Transcription elongation factor AFF2/FMR2 regulates expression of expanded GGGGCC repeat-containing C9ORF72 allele in ALS/FTD. Nat Commun 2019, 10: 5466.
Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 2013, 5: 208ra149.
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 2013, 110: E4530–4539.
Batra R, Nelles DA, Pirie E, Blue SM, Marina RJ, Wang H, et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 2017, 170: 899-912 e810.
Yamada SB, Gendron TF, Niccoli T, Genuth NR, Grosely R, Shi Y, et al. RPS25 is required for efficient RAN translation of C9orf72 and other neurodegenerative disease-associated nucleotide repeats. Nat Neurosci 2019, 22: 1383–1388.
Cheng W, Wang S, Zhang Z, Morgens DW, Hayes LR, Lee S, et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron 2019, 104: 885–898 e888.
Zhou Q, Mareljic N, Michaelsen M, Parhizkar S, Heindl S, Nuscher B, et al. Active poly-GA vaccination prevents microglia activation and motor deficits in a C9orf72 mouse model. EMBO Mol Med 2020, 12: e10919.
Nguyen L, Montrasio F, Pattamatta A, Tusi SK, Bardhi O, Meyer KD, et al. Antibody Therapy Targeting RAN Proteins Rescues C9 ALS/FTD Phenotypes in C9orf72 Mouse Model. Neuron 2020, 105: 645–662 e611.
Lynch E, Semrad T, Belsito VS, FitzGibbons C, Reilly M, Hayakawa K, et al. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis Model Mech 2019, 12: dmm039552.
Cykowski MD, Dickson DW, Powell SZ, Arumanayagam AS, Rivera AL, Appel SH. Dipeptide repeat (DPR) pathology in the skeletal muscle of ALS patients with C9ORF72 repeat expansion. Acta Neuropathol 2019, 138: 667–670.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31871023 and 31970966), the National Key Scientific R&D Program of China (2016YFC1306000), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hao, Z., Wang, R., Ren, H. et al. Role of the C9ORF72 Gene in the Pathogenesis of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Neurosci. Bull. 36, 1057–1070 (2020). https://doi.org/10.1007/s12264-020-00567-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-020-00567-7