
Abstract
As a multi-factorial degenerative disease, Parkinson’s disease (PD) leads to tremor, gait rigidity, and hypokinesia, thus hampering normal living. As this disease is usually detected in the later stages when neurons have degenerated completely, cure is on hold, ultimately leading to death due to the lack of early diagnostic techniques. Thus, biomarkers are required to detect the disease in the early stages when prevention is possible. Various biomarkers providing early diagnosis of the disease include those of imaging, cerebrospinal fluid, oxidative stress, neuroprotection, and inflammation. Also, biomarkers, alone or in combination, are used in the diagnosis and evolution of PD. This review encompasses various biomarkers available for PD and discusses recent advances in their development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a multifactorial neurodegenerative disease that involves the progressive impairment of voluntary motor control. This represents the primary clinical feature of the disease and its prevalence increases steadily with age [1]. Impairment of voluntary motor control leads to the signs and symptoms akinesia, bradykinesia, hypokinesia, postural instability, rigidity, stooped posture, and tremor at rest, which commonly present along with gait impairment, stiffness of the arms, legs, and trunk, poor balance and coordination, and bilateral vocal cord paralysis at the extreme and worsening level. These motor features are used to monitor the response to therapy and to assess progression in PD [2, 3]. These symptoms are due to the changes at different levels within the brain. The major pathological change is the progressive degeneration of neurons in the substantia nigra pars compacta, one of the nuclei constituting the basal ganglia. These neurons are involved in transmitting dopamine to another basal ganglia nucleus and the striatum. Degeneration of these neurons leads to dysfunction of the neuronal circuits that include motor cortical areas and the basal ganglia. Ultimately, at the level of an individual’s behavior, these changes result in movement abnormalities, the major manifestations of PD, affecting the individual’s quality of life [2].
Along with the selective neuronal loss in substantia nigra pars compacta, another characteristic of PD is the Lewy body inclusions that are composed mainly of α-synuclein (ASN) [4, 5], a presynaptic protein genetically and neuropathologically linked to PD [6]. Lewy bodies are abnormal insoluble fibrillary aggregates that develop inside nerve cells in PD [7]. Apart from these prominent features, familial forms of PD can be traced to the mutations in the gene for ASN and single gene mutations, along with mutations of nuclear encoded proteins leading to the autosomal and recessive forms of PD [7]. In addition to the motor features, several non-motor features have gained attention for detecting prodromal PD, such as hyposmia (olfactory deficits), visual abnormalities (oculomotor disturbances), neuropsychiatric symptoms like depression and anxiety, anhedonia, apathy, autonomic dysfunction, orthostatic hypotension, sleep disorders such as REM-sleep behavior disorder, and constipation and other dysautonomic features. Urogenital dysfunction as well as mood disorders and other neurobehavioral abnormalities have also been reported [8, 9].
Anatomical Changes in PD
In PD, susceptibility to cognitive impairment has been suggested [10]. Thus, understanding the structural changes underlying cognitive decline is essential for diagnosis and treatment of PD. Below we describe the neuroanatomical changes that aid in the diagnosis and treatment of PD [11].
Changes in the Basal Ganglia in PD
Signals from the cortex for accurate execution of voluntary movements are processed by the basal ganglia [11, 12]. The basal ganglia are mainly involved in cognitive functions and are the most affected area in PD [11, 13, 14]. There are two subtypes of PD, tremor-dominant (TD) and with postural instability/gait difficulty (PIGD), in each of which the basal ganglia are affected in a different manner [11, 15]. In the TD subtype, reduced fractional anisotropy within the substantia nigra and increased mean and radial diffusivity within the substantia nigra and globus pallidus have been reported. However, in PIGD, the microstructure within the substantia nigra is severely affected [11, 16]. Severe reduction of the neuromelanin pigmentation, neuronal loss, and Lewy bodies have been observed in the substantia nigra [11, 17]. Depending on the severity and duration of the disease, the basal ganglia undergo different degrees of dysfunction [11, 18]. Reduction in the volumes of the caudate nucleus, thalamus, and white matter is an early sign during disease progression. Since the basal ganglia are responsible for voluntary motor control, procedural learning, eye movement, and cognitive and emotional functions, all of these are hampered [11, 19].
Changes in the Cerebellum in PD
The cerebellum shows increased activity during cognition, which is impaired in PD [11, 20]. Certain PD-related morphological changes have been observed in the cerebellum, such as decreased grey matter volume in the right quadrangular lobe along with significant contraction in the left cerebellum. The cerebellum has reciprocal connections with the basal ganglia [11, 21, 22]. As the cerebellum receives dopaminergic projections from the basal ganglia, and dopaminergic receptors are also present in the cerebellum, changes in cerebellar activation are induced by degeneration of these dopaminergic neurons [11, 23, 24].
Changes in Brain Volume in PD
The cognitive functions of PD patients are strongly correlated with brain size [11, 25]. In PD patients, atrophy of the brain has been reported in many cortical and subcortical areas, which contributes to the decrease in the volume of the brain [11, 26]. Also, volumes of the frontal lobe, the temporoparietal junction, the parietal lobe, the insula, the anterior cingulate cortex, the basal ganglia, and the thalamus are increased in PD patients [11, 27].
Changes in the Thalamus in PD
Thalamic stimulation leads to the enhancement of cognition through the hippocampus and neocortex and the modulation of gene expression, whereas thalamic lesions have been found to impair cognition [11, 28, 29]. The changes in the thalamus include 30%–40% neuronal loss along with a decrease in volume [11, 19, 30]. Specific thalamic nuclei are involved in PD. Atrophy of the caudal intralaminar nucleus and hypertrophy of the other nuclei result in an altered shape of the thalamus. Depression of PD patients is due to the changes in the mediodorsal thalamus [11, 31, 32]. Also, a significant reduction in fractional anisotropy has been reported in the anterior, dorsomedial, and ventral anterior nuclei of the thalamus [11, 33].
Changes in the Hypothalamus in PD
In PD patients, impairment of hypothalamic function has been reported and the hypothalamus plays a role in cognitive functions [11, 34, 35]. Predominant degeneration has been observed in the tuberomamillary nucleus and lateral and posterior hypothalamic nuclei, along with neural degeneration in all 13 nuclei of the hypothalamus [36]. Further, decreased levels of neurotransmitters such as dopamine, melanin, serotonin, and hypocretin have been found in the hypothalamus [11, 36]. Due to the disruption in hypothalamic functioning, secondary degeneration occurs in structures such as the striatum where synthesis of dopamine occurs [11, 37]. Due to the dopamine dysfunction in the hypothalamus, sleep disorders as well as endocrine and autonomic disorders develop in PD [11, 38].
Changes in the Limbic System in PD
In PD, changes in the limbic system lead to atrophy of the grey matter which, along with the dopamine dysfunction, leads to changes in creativity and emotional dysfunction in PD patients [11, 39, 40]. Cortical, accessory basal and granular nuclei are the least affected areas whereas accessory cortical and central nuclei are more affected in PD [11, 41].
Changes in the Locus Coeruleus in PD
In PD patients, there is a significant dopaminergic neuronal loss in locus coeruleus. Due to the neuronal loss in PD, symptoms such as resting tremors occur [11, 42, 43]. In the locus coeruleus, lesions of the dopaminergic system result in metabolic dysfunction in the cerebral cortex and impairment of cognitive functions [11, 44].
Changes in Glial Cells in PD
In response to the physiological and pathological conditions, structural changes occur in astrocytes, which influence neurons through non-synaptic communication [45,46,47]. This altered neuroglial interaction is the underlying cause of neurodegenerative diseases like PD [48].
Neurotransmitter Changes in PD
In PD, the signs and symptoms develop only after dopamine deficiency is up to 80% in the basal ganglia (caudate and putamen) and the substantia nigra pars compacta. Other neurotransmitters such as acetylcholine (ACh), γ-aminobutyric acid (GABA), and glutamate also play a major role [49, 50]. In the extrapyramidal system, there is a neurotransmitter imbalance with a deficiency in dopamine and GABA and a surplus of ACh and glutamate. Dopamine and ACh act as postsynaptic excitatory neurotransmitters, through D1/D2 receptors and muscarinic and nicotinic cholinergic subreceptors, respectively [49, 51]. GABA exerts a reduced presynaptic inhibitory action and glutamate exerts an excitotoxic effect and a presynaptic inhibitory action via N-methyl-D-aspartate (NMDA) and metabotropic glutaminergic receptors [49, 51].
Dopamine
PD is pathologically characterized by a loss of dopaminergic neurons in the substantia nigra pars compacta, due to oxidative stress and partly to susceptibility genes leading to a lower dopamine level. This lowering in dopamine level further leads to characteristic motor impairments in PD. The role of dopamine in PD is supported by various studies which showed effectiveness of dopamine agonists in ameliorating the motor defects of PD [49].
Acetylcholine
In PD, there is an imbalance between dopaminergic–cholinergic neurotransmission via the D2 receptors and a surplus of muscarinic cholinergic neurons via M4 receptors. In PD caused by depletion of dopamine, the autoinhibition of acetylcholine release by auroreceptor is blocked, which results in surplus acetylecholine release. However, the dopaminergic neurons are activated partly by nicotinic cholinergic neurons [49, 52] along with the activation by nACh neurons in the putamen via beta-2 receptors. Thus, beta-2 nACh agonists could be of therapeutic value in the treatment of PD [49, 53].
Serotonin
Serotonergic (5-HT) neurons have connections to almost all brain structures and show a high degree of axonal branching. The loss of dopamine can be compensated by 5-HT hyperactivity. There is a dopaminergic and serotonergic interaction in the putamen through glutaminergic neurons via NMDA receptors and 5-HT exerts its function via 5-HT2A receptors. Additional administration of 5-HT2A antagonists not only improves dyskinesias, but also improves psychotic symptoms [49, 54].
GABA
GABA exerts a presynaptic inhibitory action in the striatum, in the globus pallidus internus which is an output nucleus, and in the globus pallidus externus which is an input nucleus, via GABAA and GABAB receptors. The dopamine deficiency is enhanced by GABA hypofunction together with antagonistic interaction between AMPA/NMDA glutaminergic neurons and GABAA receptors [49]. At present, agents that enhance the GABAergic neurotransmission with an agonistic effect particularly on GABAA receptors could be of therapeutic value for PD [49, 55].
Glutamate
In PD there is an imbalance in GABAergic-glutaminergic neurotransmitters, with a GABA deficiency and a glutamate hyperactivity or excitotoxicity in the extrapyramidal system. Glutaminergic neurons strongly presynaptically inhibit D2 dopaminergic neurons via NMDA receptors in the putamen [49, 56].
Biomarkers for Parkinson’s Disease
Currently, one of the significant limitations in the treatment of PD is late detection. Despite the rigorous efforts in patient management and clinical research, methods are suboptimal in areas of diagnosis, refining prognosis, predicting individual responses to therapeutic interventions, and tracking disease progression. Thus the need of validated biomarkers with a high degree of sensitivity and specificity is the need of the hour with the intuitive goal to help diagnose disease [57]. The following encompasses various biomarkers available for PD and discusses recent advances in their development.
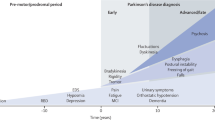
A biomarker gives an indication of the particular disease state of an organism and evaluates the progress of the disease and the treatment effects. It can be a physical, chemical, or biological parameter [58]. As per the NIH study group, it is objectively measured and evaluated as an indicator of normal biologic processes and pathogenic or pharmacologic responses to a therapeutic intervention [58]. In the diagnosis of PD, although no individual biomarkers have been recommended, they can be combined rationally to predict the status and progression of the disease. The difficulties in diagnosing PD make the search for biomarkers difficult, as identifying the diagnostic criteria for a disease is important in identifying and validating the biomarkers. Although biomarkers of PD are diverse, they can be broadly divided into three stages: Prodromal, preclinical, and clinical PD. Table 1 summarizes the biomarkers of PD at these stages.
Imaging Biomarkers
Imaging biomarkers are being increasingly employed in the diagnosis of PD to provide support for clinical observations. The imaging biomarkers include molecular imaging, transcranial sonography, magnetic resonance imaging (MRI), and optical coherence tomography (OCT).
Molecular Imaging
Molecular imaging techniques are not just those that offer anatomical images. In PD, molecular imaging is a type of medical imaging that provides pictures of the events happening inside the body at the cellular and molecular levels and measures the chemical and biological processes as compared to the other imaging techniques. In PD, it measures the functions of neurons and other brain tissues that use neurotransmitters like dopamine which become abnormal. Molecular imaging procedures are noninvasive, safe, and painless. This involves an imaging device, an imaging agent, and a probe. There are two widely used imaging techniques that focus on dopamine, dopamine transporter single-photon emission computed tomography (DAT-SPECT) and fluorodopa positron emission tomography (F-DOPA PET) [59].
DAT SPECT
DAT SPECT is an FDA-approved approach [60] to scanning for the functioning of presynaptic dopamine transporters and visualizing dopamine degeneration in the nigro-striatum in PD [61]. DaTscan (ioflupane I123 injection, also known as phenyltropane) is a radiopharmaceutical agent which is injected intravenously in a procedure called SPECT imaging. The 123I-N-3-fluoropropyl-2beta-carbomethoxy-3beta-4-iodophenyl tropane (123I-FP-CIT) binds to DAT proteins in the presynaptic membrane on the dopaminergic terminals from the substantia nigra to the striatum thus serving as a marker for dopamine terminal innervation. This depicts the DAT abnormality in PD and predicts the course of the disease by measuring the number of DATs relative to the normal level at any early point in PD [60, 61].
Fluorodopa PET Scan
F-DOPA is a fluorinated form of levodopa (L-DOPA). F-DOPA is used as a radiotracer in PET scans, which focus on analyzing the efficiency of neurons in the striatum in dopamine usage and serve as a biomarker for PD [62]. The F-DOPA scan involves a neutral amino-acid that is transported into presynaptic neurons by an active transport system and is converted into fluorodopamine by the enzyme aromatic amino-acid decarboxylase (AADC). This is a rate-limiting step during dopamine synthesis in dopaminergic neurons. [18F] DOPA uptake reflects the ability of dopaminergic neurons to produce dopamine through AADC. The fluorodopamine is stored in presynaptic vesicles until neuronal activation triggers its release, and subsequently binds to dopamine receptors and then enters catecholamine storage vesicles.
When injected into the bloodstream, 6-fluoro (18F)-L-DOPA crosses the blood-brain barrier (BBB) and ultimately reaches the dopaminergic cells. It is then used as a precursor of dopamine for the assessment of presynaptic dopaminergic integrity and accurately reflects the monoaminergic disturbances in PD. This test specifically distinguishes PD from other types of neurodegeneration. It assesses the cerebral metabolism of glucose thereby serving as a biomarker for neuronal activity and neurodegeneration. This reduced brain metabolism is depicted in the cortex, cerebellum, and striatum [62]. The current status of 18F-DOPA PET scans for PD is that 18F-DOPA is a PET tracer with selective in vivo affinity for the basal ganglia due to the specific metabolism of the substantia nigra, and that 18F-DOPA PET imaging is the best diagnostic tool for PD [63]. In a retrospective study of PD patients (n = 29) scanned with 18F-DOPA PET, an association has been established between reduced 18F-DOPA uptake and key symptoms of PD such as hypokinesia-rigidity. There is hypothesized to be a stronger correlation between dopamine depletion in the striatum with hypokinesia-rigidity than with tremor.
The results showed that reduced F-DOPA uptake is contralateral to the hypokinesia-rigidity symptoms and is also correlated with its severity. Furthermore, freezing of gait is correlated with reduced F-DOPA uptake in the putamen of the right hemisphere. But for tremor severity, no correlation has been seen with F-DOPA reduction, as tremor in PD is not only based on dopamine-related pathway but may also rely on a different pathway [64].
In another study, the visible and quantitative anatomic distribution of 18F-labelled L-DOPA in the healthy human brain was evaluated to understand the extrastriatal sites of levodopa function and the fluorodopa accumulation in patients. F-DOPA PET imaging demonstrated trapping of the labeled dopamine or its metabolites in substantial quantities in many brain areas and not specifically in dopaminergic or catecholaminergic neurons. The total uptake of florodopa is correlated with the sum of the catecholamine and indolamine concentrations in the brain. Substantial dopamine production within serotonin and norepinephrine neurons may play a role in therapeutic effects of L-DOPA [65].
Transcranial Sonography
Transcranial sonography is a non-invasive, painless technique that can be performed with no need for anesthesia [66, 67]. Transcranial sonography helps to visualize the mesencephalic hyperechoic signals in the substantia nigra pars compacta in PD that represent a dysfunction in the dopaminergic nigrostriatal pathway [67]. Hyperechogenicity is an echo that is produced due to fat deposits during the ultrasound examination of an organ. The mesencephalic substantia nigra hyperechogenicity in PD patients is believed to be associated with increased iron concentrations that result in oxidative stress and further injury to dopaminergic neurons, causing PD. Elevated nigral iron levels have been reported in PD, reflecting disturbed brain iron homeostasis [68]. Nigral iron elevation is an invariable feature of PD and is considered to be a sufficient cause of neurodegeneration in PD [69]. Neurodegeneration along with brain iron accumulation is caused by a mutation in pantothenate kinase 2 (PANK2), which catalyzes the initial step in coenzyme A synthesis. Mutations reduce the catalytic activity of PANK2 along with iron accumulation in the basal ganglia. Patients with this type of mutation often present with Parkinsonism, as the brains of affected patients contain Lewy bodies, indicating that iron accumulation is upstream of ASN deposition in idiopathic PD [69]. Changes in the echogenicity are usually due to morphological and functional changes in the PD brain [67]. Thus, this procedure is reliable in detecting abnormalities in the basal ganglia. The area of the substantia nigra has been measured using a Sonos 5500 ultrasound device. Echogenicity of the raphe, red nuclei, thalamus, caudate, lenticular nuclei, and width of the third and lateral ventricles have been documented [66].
Magnetic Resonance Imaging (MRI)
MRI imaging in PD has particularly demonstrated the presence of increased iron in the substantia nigra in Parkinsonian individuals. One of the most developed MRI markers is the iron load, using T2/T2* relaxometry [71]. Using T2 and T2* imaging of the substantia nigra in PD patients, a change in the relaxation time constants can be measured as a substitute for increased iron concentration in PD. These brain changes in PD have been found with a powerful MRI (3 Tesla scanner) that generates a magnetic field twice the strength of 1.5 Tesla machines and 10-to-15 times the strength of open MRI scanners [70]. Further, the T2 relaxation time also demonstrates the speed of water magnetization and its return to equilibrium after perturbation by a radiofrequency pulse during this MRI scan. T2* imaging and its reciprocal R2* are essential in nigral imaging and demonstrate the macroscopic nigral changes in iron [70]. This method mainly reflects non-heme iron rather than heme-iron. These imaging techniques are more sensitive to bound iron stored as ferritin or neuromelanin [70].
MRI has become a standard technique that is routinely performed in patients with PD in order to exclude secondary causes and provide specific information that aids in the diagnosis of a neurodegenerative disease like PD. Transcranial sonography is a more widely used and a more recent advance in the diagnosis of PD. It may detect individuals in the premotor phase of PD, and from this perspective transcranial sonography is considered superior to MRI [72].
Optical Coherence Tomography (OCT) as a Recent Biomarker for PD
OCT provides accurate cross-sectional imaging of the internal structures in biological tissues to reveal various inner retinal or optic nerve pathologies. Vision is one of the non-motor systems that are altered in PD. Neurochemical analysis of the eyes of patients with PD has shown decreased retinal dopamine concentration, resulting in decreased visual acuity. In the human retina, dopamine is released by a set of amacrine cells in the inner proximal layer of the retina that communicate with other cells to play a role in channeling visual information through the retina. Thus the thickness of the retinal nerve fiber layer is a potential biomarker for PD diagnosis [73]. Retinal assessment is performed by DARC (detection of apoptotic retinal cells) and by OCT [74].
Biochemical Biomarkers
Biochemical biomarkers can be investigated either in the cerebrospinal fluid (CSF) or in blood. CSF is an accessible source of brain-derived proteins [75]. It is separated by the BBB and supplies the brain tissues with nutrients and filters waste from the brain interstitial fluid. In PD, there are disruptions in the BBB and thus the CSF can be investigated for potential biomarkers [76]. Also, the CSF remains in close contact with the extracellular space of the brain. Proteins and peptides reflective specifically of brain-derived activities can diffuse into the CSF and many of the biochemical modifications would be reflected in the CSF. Thus CSF may be a potential biomarker [76]. Many of the CSF proteins basically originate from the blood rather than directly from the surrounding brain tissue. CSF samples are usually obtained by lumbar puncture in the L3-4 or L4-5 interspace [77]. Following are some of the potential biochemical biomarkers for early detection of PD.
Glial Fibrillary Acidic Protein (GFAP)
Astrocytes are the most abundant cell type in the human central nervous system [78]. Apart from being central to the catabolism of specific amino-acids, they are used in the synthesis of new amino-acids in the brain [79]. Astrocytes are essentially involved in brain metabolism and the transport of multiple nutrients and metabolic precursors to neurons via the malate-aspartate shuttle and other transporters. Protoplasmic astrocytes envelop neuronal cell bodies and synapses beside an increase in the accumulation of ASN, whereas fibrous astrocytes interact with the nodes of Ranvier and oligodendroglia [78, 80]. Astrocytes are characterized by the expression of the intermediate filaments GFAP and vimentin [78]. GFAP is the major intermediary filament of astrocytes [80, 81]. It is a highly specific brain protein involved in maintaining the shape and motility of the astrocytic processes. It also contributes to white matter architecture, BBB integrity, and myelination [81]. Extra-cerebral sources of GFAP have not yet been identified and its blood levels in healthy individuals are very low. Release of GFAP from brain tissue into the blood stream is supposed to occur under these conditions: disruption of the BBB, and the loss of astrocytic structural integrity due to necrosis or mechanical disruption. It is also a key intermediate filament 3 protein responsible for maintaining the structure of glia and maintaining their mechanical strength as well as supporting neighboring neurons and the BBB [82]. Evidence has suggested that GFAP and its breakdown products (GFAP-BDPs) serve as a biofluid-based biomarker for neurological conditions like PD. Injury to the astrocytes causes the release of GFAP-BDPs and to a lesser extent, full-length GFAP, from the injured astrocytes to the interstitial/extracellular fluid. They are then released into the circulating blood by direct venous drainage (lymphatic pathway) or further continue to follow the CSF flow and eventually enter the circulation by diffusing through the BBB. One of the major advantages of GFAP as a biomarker is that it has a strong brain-specificity and is highly expressed in the brain [83, 84].
Astrocytic functions become transiently impaired after astrocytic damage and this impacts the neurons and leads to PD [78]. Thus, high levels of CSF GFAP and blood-serum GFAP are a potential early biomarker for neurological disorders such as PD. The GFAP level in CSF is usually measured with an in-house ELISA kit based on polyclonal antibodies, with a lower limit of 70 ng/L and intra- and inter-assay coefficients of variation of 4% and 8%, respectively [81].
DJ-1
DJ-1, also known as Parkinsonism-associated deglycase encoded by the PARK7 gene [85], is a multifunctional protein that plays a neuroprotective role in oxidative stress during neurodegeneration, apart from its other functions of transcriptional regulation, antioxidative stress reaction, and chaperone, protease, and mitochondrial regulation [85, 86]. DJ-1 is usually expressed throughout the body in almost all the cells and brain tissues in both neurons and glia [85]. Specifically, the expression of DJ-1 is increased in oxidative stress. An increase of DJ-1 ultimately reflects oxidative stress in PD patients, making it a biomarker for PD [28, 29]. However, it should be noted that as the DJ-1 level is very high in erythrocytes, hemolysis and contamination by erythrocytes greatly affect the DJ-1 levels in CSF and plasma [85]. Therefore, in determination of the DJ-1 content in plasma and CSF, an evaluation of hemolysis and contamination by erythrocytes is necessary [85].
Urate
Urate is the anionic form of uric acid (2, 6, 8-trioxy-purine) that predominates at neutral pH both intracellularly and in all body fluids. Urate is synthesized by xanthine oxidoreductase through successive oxidization of hypoxanthine to xanthine and then to urate [87]. Uric acid is an important endogenous antioxidant with high concentrations in brain and serum. It can prevent oxidative stress due to its ability to scavenge reactive oxygen species (ROS) and reactive nitrogen species (RNS) [79, 87, 88].
Evidence from meta-analysis results has shown a lower serum level of uric acid in patients with PD than in healthy controls, and this finding is more marked in men than in women. Thus the serum uric acid level may be a potential biomarker for PD [87].
Brain-Derived Neurotrophic Factor (BDNF)
BDNF is a potent inhibitor of apoptosis-mediated cell death and the neurotoxin-induced degeneration of dopaminergic neurons. Thus, the BDNF is eventually used in the development of neuroprotective therapies to improve cognitive functioning in PD [89,90,91]. Implicated in regulating neuronal survival, the reduced expression of BDNF within the substantia nigra is associated with the deterioration of dopaminergic neurons. Thus, BDNF serves as a potential biomarker in PD. The CSF level of BDNF is correlated with cognitive performance in PD patients. BDNF levels can be assessed with the ELISA method [89].
Neurofilament Light Chain Protein
Neurofilaments are major structural elements involved in maintaining neuronal shape and size and axonal caliber. Thus, they are necessary for maintaining neuronal integrity and the conduction of nerve impulses along the axon. Neurofilaments comprise three subunits [92, 93], neurofilament light (60 kDA), neurofilament medium (100 kDA), and neurofilament heavy (110 kDA) chain proteins. The presence of neurofilament light chain proteins in the CSF is indicative of neuronal degeneration. In particular, abnormally phosphorylated neurofilaments have been identified in PD associated with Lewy bodies. Thus, neurofilaments in the CSF can serve as a potential biomarker to predict the extent of neurodegeneration. Elevated levels of neurofilaments have been detected in CSF following axonal damage in the CNS that correlates with age. Increased levels of neurofilaments reflect degeneration of large myelinated axons. This can be useful in the differential diagnosis of PD, assessing the increasing levels of NFL as this is very sensitive in detecting more aggressive neuronal death [92, 93].
Glutathione
It is well known that redox stress contributes to PD progression [95] and is involved in the degeneration of dopaminergic neurons. Disruption of physiological maintenance of the redox potential in neurons interferes with several biological processes and ultimately leads to cell death. Glutathione functions as an antioxidant that clears out free radicals that could damage and potentially be toxic to dopaminergic neurons, thus operating as a neuroprotective agent. The total glutathione comprises reduced (GSH) and oxidized forms, maintains redox homeostasis, clears metabolic waste, and serves as a reservoir for amino-acids in the brain [94].
Post-mortem analysis of nigral tissue from PD patients has revealed deficiency of GSH, indicating an impaired ability of cells to metabolize cellular waste and defense against ROS, RNS and H2O2 [94].
Coenzyme Q10
Coenzyme Q10 has recently gained attention as a dietary supplement. It is present in the cytosol and plasma in various neurodegerative diseases including PD. Coenzyme Q10 is an essential co-factor in the mitochondrial respiratory chain and in oxidative phosphorylation, and is considered to be a relevant antioxidant in PD, as it helps in the functioning of the mitochondrial transport chain. In vitro and in vivo studies have suggested a defect in mitochondrial complex 1 that results in the disruption of redox equilibrium, which ultimately leads to neuronal toxicity. As a lipophilic antioxidant, it scavenges the radicals within membranes. A decreased concentration of this coenzyme results in an increase of free radicals not being scavenged, leading to neuronal degeneration [96]. Thus coenzyme Q10 significantly retards the progression of PD, and is clinically available as a peripheral biomarker as it is capable of identifying diminished coenzyme Q10 activity and has the potential to enhance the clinical outcome in PD [96].
Neuromelanin
Neuromelanin is a dark polymer pigment present in catecholaminergic neurons and it appears to be abundant in the human brain but is absent from the brains of many lower species [97, 98]. In PD, cells in the substantia nigra that contain neuromelanin are affected [98]. Accumulation of neuromelanin-containing neurons seems to be a protective phenomenon that prevents various neurotoxic processes. In dopaminergic neurons of the substantia nigra where no ferritin has been detected, neuromelanin functions as an iron storage system. In PD, dying neurons release neuromelanin and this triggers a vicious cycle of neuroinflammation which ultimately leads to neuronal death [96, 98]. Neuromelanin also protects neurons from oxidative stress mediated by free radicals and metals [99]. The neuromelanin level can be measured by MRI techniques [90, 100], and it has been demonstrated to provide information on substantia nigra degeneration and is an essential biomarker for PD.
Plasma Homocysteine
Homocysteine is a naturally occurring amino-acid resulting from the methylation process. The homocysteine levels are influenced by folate concentrations and several genetic factors. An elevated concentration of total homocysteine in plasma and CSF is considered to be a risk factor for PD [101]. Homocysteine triggers neuroinflammation and activated astrocytes and microglia release a number of factors that further trigger inflammatory responses, ultimately leading to neuronal death [102]. Also, homocysteine causes neuroinflammation, leading to NO release from the support cells of the nervous system. This can trigger the release of NO within neurons themselves, triggering apoptosis [102, 103].
α-Synuclein and Other Lysosomal Enzymes
Lysosomal dysfunction and impairment is increasingly recognized as a central event in the pathophysiology of PD, thus serving as a potential biomarker for PD. Lysosomes are a part of the cellular waste-disposal system and their dysfunction is involved in PD pathogenesis. Apparently this autophagy-lysosomal system is responsible for the hydrolysis of dysfunctional proteins, and in the case of lysosomal impairment, ASN aggregation is observed as a common hallmark of PD. This ASN aggregation also involves post-translational modifications and certain unknown factors. Certain lysosomal enzymes such as glucocerebrosidase and cathepsin have been reported in lysosomal dysfunction. Consequently, detection of the lysosomal enzymes and proteins in the CSF could serve as a biomarker for PD. However, CSF lysosomal enzyme activity alone cannot discriminate PD from other diseases. The combination of CSF lysosomal markers with ASN species and indicators of mitochondrial dysfunction, inflammation, and other pathological proteins in PD may facilitate a more accurate diagnosis [80]. In addition, deficiency of the lysosomal enzyme beta-glucocerebrosidase (GCase) increases the risk of Parkinsonism, which appears to be driven by a direct effect of GCase deficiency and lysosomal dysfunction on α-synuclein aggregation. Therefore, the GCase activity in the CSF would further serve as a PD biomarker [80].
Inflammatory Biomarkers
Neuroinflammatory reactions are involved in idiopathic PD [85] and this process occurs alongside the loss of dopaminergic neurons in PD [87]. Microglial cells are one of the major cell types involved in the inflammatory process in the CNS [86]. Under physiological conditions, the quiescent state of microglia is maintained by a variety of immunomodulators such as CX3CL1, CD200, and neural cell adhesion molecule. The CX3CL1–CX3CR1 signaling negatively regulates microglial activation, thus protecting the DA neurons from degeneration by neurotoxins. Deficiency of this signaling results in increased neurotoxicity and enhanced cell death of dopamine neurons in the substantia nigra pars compacta of animal PD models. Dysfunction of CD200–CD200R signaling also increases the activation of microglia and exacerbates the degeneration of dopamine neurons in rat PD models, leading to neuroinflammation [86].
One inducer through which neurons and the microglia communicate to regulate inflammation is fractalkine (CX3CL1). Fractalkine is a 373-amino-acid protein that is secreted by neurons and exists in both membrane-bound and soluble forms. The membrane-bound form serves as an adhesion molecule to support leukocyte adhesion. The soluble form functions as a pro-inflammatory chemoattractant and serves as an anti-inflammatory and neuroprotective agent that reduces neuronal apoptosis. Thus, low levels of fractalkine contribute to neuroinflammation, leading to neurodegeneration in PD [104]. Thus fractalkine levels are associated with disease severity and the progression of PD [104].
Neurosin
Neurosin is a trypsin-type serine protease that is preferentially expressed in the human brain, and can be identified by northern blotting. Neurosin is a protease that cleaves [66] and degrades ASN, the major constituent of Lewy bodies present in the brain of patients with synucleinopathies like PD [88]. In synucleinopathies such as PD, lower neurosin levels have been reported [66]. The potential link between neurosin and its substrate ASN has been investigated in vivo using a commercial sandwich ELISA kit and a direct ELISA developed in-house to quantify CSF levels of neurosin and ASN in patients with PD [88, 105].
Clinical Biomarkers
In PD, several clinical biomarkers show motor impairment symptoms such as essential tremors, postural irregularity, bradykinesia, muscular rigidity, walking difficulty, incontinence, muscle rigidity, falling, and drooling. The PaRkInson And non-Motor symptOms (PRIAMO) study, which was conducted to evaluate non-motor symptom prevalence in PD, showed that in the early stage patients had psychiatric disturbances, fatigue, and attention and memory problems [106]. Along with the non-motor symptoms such as nocturia, altered circadian rhythm, hyposmia, hypoguesia, impaired visuo-spatial and color discrimination, cardiac sympathetic innervation, reduction in the sympathetic skin response, neurobehavioral deficits like depression, dementia, olfaction, nocturia, impaired circadian rhythm, narcolepsy, and impulsive behavior are clinical biomarkers [107]. These biomarkers are helpful and important for assessing the effect of treatment.
Motor Function as a Clinical Biomarker
Epidemiological studies suggest that a large proportion of patients with essential tremors also have PD [108]. Further, many PD patients show pre-existing essential tremors. Complex skills like visually-guided movement, writing, velocity of movement, or reaction time are used clinically for examination.
Psychological and Affective Biomarkers
In early PD there are reports of depression, sleep disorder, apathy, and anxiety [109]. Although there are many reports of such conditions in the early stage of PD, they are not specific and need to be seen in combination with other biomarkers [109]. Several studies have reported sleep disturbances with rapid eye movement disorder in PD patients [110].
Olfactory Dysfunction
There is evidence of olfactory dysfunction in PD. In a large proportion of PD patients, hyposmia and anosmia occur accompanied by loss of smell detection and identification [111]. This suggests neurodegeneration in the olfactory bulb. Combined with neuroimaging and neurochemical markers, olfactory testing can be a good tool for early detection of PD [112].
Vision Disturbance
Since dopaminergic pathways are associated with the control of oculomotor function, impairment of saccades has been observed in early PD. However due to intra-subject variability, this biomarker has low reproducibility [113].
Genetic Biomarkers
Single genes leading to the heritable forms of PD have yet to be identified. Analysis for mutations in SNCA, Parkin, PINK1, DJ1, LRRK2 and GBA is of utmost importance [103, 114]. The SNCA gene encodes the ASN protein and corresponds to the PARK1 and PARK4 loci and mutations in SNCA gene account for >1% in the general population [114, 115]. SNCA encoding ASN, the principal component of Lewy bodies, is the cause of PD [103, 116]. Triplication of SNCA gene causes a two-fold increase in ASA expression. In PD patients, triplication of this gene may also cause orthostatic hypotension. In 2008 one such case of duplication was reported in which the patient showed no response to levodopa treatment. Genome expression analysis can be performed using peripheral blood leucocytes and PD patients compared to healthy controls [117]. Analysis of global gene expression with DNA microarrays has been performed in the peripheral blood of PD patients [117, 118].
Conclusion
PD is one of the leading neurodegenerative disorders and most of the cardinal symptoms appear late. By that time, most of the dopaminergic neurons are already damaged. Thus, it has become important to identify biomarkers that can detect the disease early. Biomarkers are important for improving diagnosis and are helpful in monitoring disease progression. For better understanding of the progression of disease, biomarkers in PD should be sensitive and specific. Investigations for new biomarkers are restricted by many challenges such as the limited availability of specimens for validation studies. Discovery of new biomarkers can play a significant role in the diagnosis and treatment of PD. These preclinical biomarkers will play an essential role in identifying individuals at-risk of PD. Thus current PD research demands a search for unique biomarkers that can be useful to discriminate between PD and other diseases with higher sensitivity and specificity and preferably one such biomarker that can entirely track the progression in PD.
References
Frisardi V, Santamato A, Cheeran B. Parkinson’s disease: new insights into pathophysiology and rehabilitative approaches. Parkinson’s Dis 2016, 2016: 3121727.
Mazzoni P, Shabbott B, Cortés JC. Motor control abnormalities in Parkinson’s disease. Cold Spring Harb Perspect Med 2012, 2: a009282.
Teive HA, Bertucci Filho DC, Munhoz RP. Unusual motor and non-motor symptoms and signs in the early stage of Parkinson’s disease. Arquivos de Neuro-Psiquiatria 2016, 74: 781–784.
Miki Y, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K. PLA2G6 accumulates in Lewy bodies in PARK14 and idiopathic Parkinson’s disease. Neurosci Lett 2017, 645: 40–45.
Gupta R, Kim C, Agarwal N, Lieber B, Monaco EA. Understanding the influence of Parkinson disease on Adolf Hitler’s decision-making during World War II. World Neurosurg 2015, 84: 1447–1452.
Song LK, Ma KL, Yuan YH, Mu Z, Song XY, Niu F, et al. Targeted overexpression of α-synuclein by rAAV2/1 vectors induces progressive nigrostriatal degeneration and increases vulnerability to MPTP in mouse. PLoS One 2015, 10: e0131281.
Cieri D, Brini M, Calì T. Emerging (and converging) pathways in Parkinson’s disease: keeping mitochondrial wellness. Biochem Biophys Res Commun 2017, 483: 1020–1030.
Wu Y, Le W, Jankovic J. Preclinical biomarkers of Parkinson disease. Arch Neurol 2011, 68: 22–30.
Mahlknecht P, Seppi K, Poewe W. The concept of prodromal Parkinson’s disease. J Parkinson’s Dis 2015, 5: 681–697.
Solari N, Bonito-Oliva A, Fisone G, Brambilla R. Understanding cognitive deficits in Parkinson’s disease: lessons from preclinical animal models. Learn Mem 2013, 20: 592–600.
Prakash KG, Bannur BM, Chavan MD, Saniya K, Sailesh KS, Rajagopalan A. Neuroanatomical changes in Parkinson’s disease in relation to cognition: An update. J Adv Pharm Technol Res 2016, 7: 123
Blandini F, Nappi G, Tassorelli C, Martignoni E. Functional changes of the basal ganglia circuitry in Parkinson’s disease. Prog Neurobiol 2000, 62: 63–88.
Brown LL, Schneider JS, Lidsky TI. Sensory and cognitive functions of the basal ganglia. Curr Opin Neurobiol 1997, 7: 157–63
Obeso JA, Rodríguez-Oroz MC, Benitez-Temino B, Blesa FJ, Guridi J, Marin C, et al. Functional organization of the basal ganglia: Therapeutic implications for Parkinson’s disease. Mov Disord 2008, 23(Suppl 3): S548–559
Jankovic J, McDermott M, Carter J, Gauthier S, Goetz C, Golbe L, et al. Variable expression of Parkinson’s disease: A base-line analysis of the DATATOP cohort. The Parkinson Study Group. Neurology 1990, 40: 1529–1534.
Nagae LM, Honce JM, Tanabe J, Shelton E, Sillau SH, Berman BD. Microstructural changes within the basal ganglia differ between Parkinson disease subtypes. Front Neuroanat 2016, 10: 17.
Dickson DV. Neuropathology of movement disorders. In: Tolosa E, Jankovic JJ (Ed.). Parkinson’s Disease and Movement Disorders. Hagerstown, MD: Lippincott Williams & Wilkins 2007: 271–283.
Reetz K, Gaser C, Klein C, Hagenah J, Büchel C, Gottschalk S, et al. Structural findings in the basal ganglia in genetically determined and idiopathic Parkinson’s disease. Mov Disord 2009, 24: 99–103.
Lee SH, Kim SS, Tae WS, Lee SY, Choi JW, Koh SB, et al. Regional volume analysis of the Parkinson disease brain in early disease stage: Gray matter, white matter, striatum, and thalamus. AJNR Am J Neuroradiol 2011, 32: 682–687.
Stoodley CJ. The cerebellum and cognition: Evidence from functional imaging studies. Cerebellum 2012, 11: 352–365.
Rolland AS, Herrero MT, Garcia-Martinez V, Ruberg M, Hirsch EC, François C. Metabolic activity of cerebellar and basal ganglia-thalamic neurons is reduced in Parkinsonism. Brain 2007, 130(Pt 1): 265–275.
Borghammer P, Østergaard K, Cumming P, Gjedde A, Rodell A, Hall N, et al. A deformation-based morphometry study of patients with early-stage Parkinson’s disease. Eur J Neurol 2010, 17: 314–20.
Wu T, Hallett M. The cerebellum in Parkinson’s disease. Brain 2013, 136(Pt 3): 696–709.
Giompres P, Delis F. Dopamine transporters in the cerebellum of mutant mice. Cerebellum 2005, 4: 105–111
Rushton JP, Ankney CD. Whole brain size and general mental ability: A review. Int J Neurosci 2009, 119: 691–731.
Watts RL, Standaertt DG, Obeso JA. Movement Disorders. 3rd ed. New York: McGraw Hill, 2011.
Cerasa A, Messina D, Pugliese P, Morelli M, Lanza P, Salsone M, et al. Increased prefrontal volume in PD with levodopa-induced dyskinesias: A voxel-based morphometry study. Mov Disord 2011, 26: 807–812.
Aglioti S. The role of the thalamus and basal ganglia in human cognition. J Neurolinguistics 1997, 10: 255–265.
Shirvalkar P, Seth M, Schiff ND, Herrera DG. Cognitive enhancement with central thalamic electrical stimulation. Proc Natl Acad Sci U S A 2006, 103: 17007–17012.
Halliday GM. Thalamic changes in Parkinson’s disease. Parkinsonism Relat Disord 2009, 15(Suppl 3): S152–155.
McKeown MJ, Uthama A, Abugharbieh R, Palmer S, Lewis M, Huang X. Shape (but not volume) changes in the thalami in Parkinson disease. BMC Neurol 2008, 8: 8.
Li W, Liu J, Skidmore F, Liu Y, Tian J, Li K. White matter microstructure changes in the thalamus in Parkinson disease with depression: A diffusion tensor MR imaging study. AJNR Am J Neuroradiol 2010, 31: 1861–1866.
Planetta PJ, Schulze ET, Geary EK, Corcos DM, Goldman JG, Little DM, et al. Thalamic projection fiber integrity in de novo Parkinson disease. AJNR Am J Neuroradiol 2013, 34: 74–79.
Zimmerman D. Thinking with your hypothalamus. Philos Phenomenol Res 2001, 63: 521–541.
Langston JW, Forno LS. The hypothalamus in Parkinson disease. Ann Neurol 1978, 3: 129–33.
Politis M, Piccini P, Pavese N, Koh SB, Brooks DJ. Evidence of dopamine dysfunction in the hypothalamus of patients with Parkinson’s disease: An in vivo 11C-raclopride PET study. Exp Neurol 2008;214: 112–116.
Sandyk R, Iacono RP, Bamford CR. The hypothalamus in Parkinson disease. Ital J Neurol Sci 1987, 8: 227–234.
Breen DP, Nombela C, Vuono R, Jones PS, Fisher K, Burn DJ, et al. Hypothalamic volume loss is associated with reduced melatonin output in Parkinson’s disease. Mov Disord 2016, 31: 1062–1066.
Xia J, Miu J, Ding H, Wang X, Chen H, Wang J, et al. Changes of brain gray matter structure in Parkinson’s disease patients with dementia. Neural Regen Res 2013, 8: 1276–1285.
Kulisevsky J, Pagonabarraga J, Martinez-Corral M. Changes in artistic style and behaviour in Parkinson’s disease: Dopamine and creativity. J Neurol 2009, 256: 816–819.
Braak H, Braak E, Yilmazer D, de Vos RA, Jansen EN, Bohl J, et al. Amygdala pathology in Parkinson’s disease. Acta Neuropathol 1994, 88: 493–500.
Isaias IU, Marzegan A, Pezzoli G, Marotta G, Canesi M, Biella GE, et al. A role for locus coeruleus in Parkinson tremor. Front Hum Neurosci 2012, 5: 179.
Bertrand E, Lechowicz W, Szpak GM, Dymecki J. Qualitative and quantitative analysis of locus coeruleus neurons in Parkinson’s disease. Folia Neuropathol 1997, 35: 80–86.
Schwartz WJ, Sharp FR, Gunn RH, Evarts EV. Lesions of ascending dopaminergic pathways decrease forebrain glucose uptake. Nature 1976, 261: 155–157.
Theodosis DT, MacVicar B. Neurone-glia interactions in the hypothalamus and pituitary. Trends Neurosci 1996, 19: 363–367.
Theodosis DT, Poulain DA, Oliet SH. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol Rev 2008, 88: 983–1008.
Villalba RM, Smith Y. Neuroglial plasticity at striatal glutamatergic synapses in Parkinson’s disease. Front Syst Neurosci 2011, 5: 68.
Voronkov DN, Khudoerkov RM, Dovedova EL. Changes in neuroglial interactions in nigrostriatal brain structures on modeling of dopamine system dysfunction. Neurosci Behav Physiol 2014, 44: 1073–1077.
Alexander GE. Biology of Parkinson’s disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin Neurosci 2004, 6: 259–280.
Varanese S, Birnbaum Z, Rossi R, Di Rocco A. Treatment of advanced Parkinson’s disease. Parkinson’s Dis 2011, 2010.
Werner FM, Covenas R. Classical neurotransmitters and neuropeptides involved in Parkinson’s disease: focus on anti-Parkinsonian drugs. Curr Drug Ther 2015, 10: 66-81
Aosaki T, Miura M, Suzuki T, Nishimura K, Masuda M. Acetylcholine–dopamine balance hypothesis in the striatum: An update. Geriatr Gerontol Int 2010, 10(s1): S148–157.
Huang LZ, Grady SR, Quik M. Nicotine reduces L-DOPA-induced dyskinesias by acting at β2* nicotinic receptors. J Pharmacol Exp Ther 2011, 338: 932–941.
Bédard C, Wallman MJ, Pourcher E, Gould PV, Parent A, Parent M. Serotonin and dopamine striatal innervation in Parkinson’s disease and Huntington’s chorea. Parkinsonism Relat Disord 2011, 17: 593–598.
Jaeger D, Kita H. Functional connectivity and integrative properties of globus pallidus neurons. Neuroscience 2011, 198: 44–53.
Werner FM, Coveñas R. Classical neurotransmitters and neuropeptides involved in generalized epilepsy: a focus on antiepileptic drugs. Curr Med Chem2011, 18: 4933–4948.
Delenclos M, Jones DR, McLean PJ, Uitti RJ. Biomarkers in Parkinson’s disease: Advances and strategies. Parkinsonism Relat Disord 2016, 22: S106–110.
Sharma S, Moon CS, Khogali A, Haidous A, Chabenne A, Ojo C, et al. Biomarkers in Parkinson’s disease (recent update). Neurochem Int 2013, 63: 201–229.
Politis M. Neuroimaging in Parkinson disease: from research setting to clinical practice. Nat Rev Neurol 2014, 10: 708–722.
Oravivattanakul S, Benchaya L, Wu G, Ahmed A, Itin I, Cooper S, et al. Dopamine transporter (DaT) scan utilization in a movement disorder center. Mov Disord Clin Pract, 3: 31–35.
Seifert KD, Wiener JI. The impact of DaTscan on the diagnosis and management of movement disorders: A retrospective study. Am J Neurodegener Dis 2013, 2: 29–34.
Niccolini F, Politis M. A systematic review of lessons learned from PET molecular imaging research in atypical parkinsonism. Eur J Nucl Med Mol Imaging 2016, 43: 2244–2254.
Calabria FF, Calabria E, Gangemi V, Cascini GL. Current status and future challenges of brain 18 imaging with F-DOPA PET for movement disorders. Hell J Nucl Med 2016, 19: 33–41.
Pikstra AR, van der Hoorn A, Leenders KL, de Jong BM. Relation of 18-F-Dopa PET with hypokinesia-rigidity, tremor and freezing in Parkinson’s disease. NeuroImage 2016, 11: 68–72.
Brown WD, Taylor MD, Roberts AD, Oakes TR, Schueller MJ, Holden JE, et al. FluoroDOPA PET shows the nondopaminergic as well as dopaminergic destinations of levodopa. Neurology 1999, 53: 1212.
Školoudík D, Jelínková M, Blahuta J, Čermák P, Soukup T, Bártová P, et al. Transcranial sonography of the substantia nigra: digital image analysis. Am J Neuroradiol 2014, 35: 2273–2278.
Bouwmans AE, Vlaar AM, Mess WH, Kessels A, Weber WE. Specificity and sensitivity of transcranial sonography of the substantia nigra in the diagnosis of Parkinson’s disease: prospective cohort study in 196 patients. BMJ Open 2013, 3: e002613.
Sian‐Hülsmann J, Mandel S, Youdim MB, Riederer P. The relevance of iron in the pathogenesis of Parkinson’s disease. J Neurochem 2011, 118: 939–957.
Ayton S, Lei P. Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. Biomed Res Int 2014, 2014.
Badea L, Onu M, Wu T, Roceanu A, Bajenaru O. Nonreproducible connectome changes hint at functional heterogeneity of Parkinson’s Disease. arXiv preprint arXiv: 1611.04794 2016.
Pyatigorskaya N, Gallea C, Garcia-Lorenzo D, Vidailhet M, Lehericy S. A review of the use of magnetic resonance imaging in Parkinson’s disease. Ther Adv Neurol Disord 2014, 7: 206–20.
Berg D, Steinberger JD, Warren Olanow C, Naidich TP, Yousry TA. Milestones in magnetic resonance imaging and transcranial sonography of movement disorders. Mov Disord 2011, 26: 979–992.
Satue M, Obis J, Rodrigo MJ, Otin S, Fuertes MI, Vilades E, et al. Optical coherence tomography as a biomarker for diagnosis, progression, and prognosis of neurodegenerative diseases. J Ophthalmol 2016, 2016: 8503859.
Normando EM, Davis BM, De Groef L, Nizari S, Turner LA, Ravindran N, et al. The retina as an early biomarker of neurodegeneration in a rotenone-induced model of Parkinson’s disease: evidence for a neuroprotective effect of rosiglitazone in the eye and brain. Acta Neuropathol Commun 2016, 4: 86.
Parnetti L, Castrioto A, Chiasserini D, Persichetti E, Tambasco N, El-Agnaf O, et al. Cerebrospinal fluid biomarkers in Parkinson disease. Nat Rev Neurol 2013, 9: 131–140.
Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Agundez JA. Cerebrospinal fluid biochemical studies in patients with Parkinson’s disease: toward a potential search for biomarkers for this disease. Front Cell Neurosci 2014, 11: 8.
Hall S, Surova Y, Öhrfelt A, Zetterberg H, Lindqvist D, Hansson O. CSF biomarkers and clinical progression of Parkinson disease. Neurology 2015, 84: 57–63.
Cabezas R, Avila MF, Torrente D, El-Bachá RS, Morales L, Gonzalez J, et al. Astrocytes role in Parkinson: a double-edged sword. Neurodegener Dis 2013, 10(5772): 54305.
Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2006, 2: 679–689.
Magdalinou N, Lees AJ, Zetterberg H. Cerebrospinal fluid biomarkers in parkinsonian conditions: an update and future directions. J Neurol Neurosurg Psychiatry 2014, 85: 1065–1075.
Mayer CA, Brunkhorst R, Niessner M, Pfeilschifter W, Steinmetz H, Foerch C. Blood levels of glial fibrillary acidic protein (GFAP) in patients with neurological diseases. PLoS One 2013, 8: e62101.
Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Agundez JA. Cerebrospinal fluid biochemical studies in patients with Parkinson’s disease: toward a potential search for biomarkers for this disease. Front Cell Neurosci 2014, 11: 8.
Yang Z, Wang KK. Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci 2015, 38: 364–374.
Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, et al. Biomarkers of traumatic injury are transported from brain to blood via the glymphatic system. J Neurosci 2015, 35: 518–526.
Saito Y. Oxidized DJ-1 as a possible biomarker of Parkinson’s disease. J Clin Biochem Nutr 2014, 54: 138–144.
Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SM. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longev 2013, 2013: 683920.
Shen L, Ji HF. Low uric acid levels in patients with Parkinson’s disease: evidence from meta-analysis. BMJ Open 2013, 3: e003620.
Tian Y, Chen K, Xie Z, Fang Y, Wang H, Nie Y, et al. The association between serum uric acid levels, metabolic syndrome and cardiovascular disease in middle aged and elderly Chinese: results from the DYSlipidemia International Study. BMC Cardiovasc Disord 2015, 15: 66
Costa A, Peppe A, Carlesimo GA, Zabberoni S, Scalici F, Caltagirone C, et al. Brain-derived neurotrophic factor serum levels correlate with cognitive performance in Parkinson’s disease patients with mild cognitive impairment. Front Behav Neurosci 2015, 9: 253.
Wennström M, Surova Y, Hall S, Nilsson C, Minthon L, Boström F, et al. Low CSF levels of both α-synuclein and the α-synuclein cleaving enzyme neurosin in patients with synucleinopathy. PLoS One 2013, 8: e53250
Marti G, Saez N, Corominas M, Cuberas G, Lorenzo C, De Fabregues O, et al. Nigrostriatal degeneration and serum Bdnf levels in patients with “de novo” untreated Parkinson’s disease. Mov Disord 2016, 31: S402.
Khalil H, Alomari MA, Khabour OF, Al-Hieshan A, Bajwa JA. Circulatory levels of Bdnf correlate with cognitive deficits in people with Parkinson’s disease. Mov Disord 2016, 31: S458.
Braissant O. Neurofilament proteins in brain diseases. In: Arlen RK (Ed.). New Research on Neurofilament Proteins. Nova Science Publishers 2007: 25–51.
Vågberg M, Norgren N, Dring A, Lindqvist T, Birgander R, Zetterberg H, et al. Levels and age dependency of neurofilament light and glial fibrillary acidic protein in healthy individuals and their relation to the brain parenchymal fraction. PLoS One 2015, 10: e0135886.
Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinson’s Dis 2013, 3: 461–491.
Mischley LK, Standish LJ, Weiss NS, Padowski JM, Kavanagh TJ, White CC, et al. Glutathione as a biomarker in Parkinson’s disease: associations with aging and disease severity. Oxid Med Cell Longev 2016, 2016: 9409363.
Mischley LK, Allen J, Bradley R. Coenzyme Q10 deficiency in patients with Parkinson’s disease. J Neurol Sci 2012, 318: 72–75.
Fedorow H, Tribl F, Halliday G, Gerlach M, Riederer P, Double KL. Neuromelanin in human dopamine neurons: comparison with peripheral melanins and relevance to Parkinson’s disease. Prog Neurobiol 2005, 75: 109–124.
Greco G. Neuromelanin and Parkinson’s disease. In: Kostrzewa RM (Ed.). Handbook of Neurotoxicity. New York: Springer, 2014: 913–932.
Lehericy S, Sharman MA, Dos Santos CL, Paquin R, Gallea C. Magnetic resonance imaging of the substantia nigra in Parkinson’s disease. Mov Disord 2012, 27: 822–830.
Nakamura K, Sugaya K. Neuromelanin-sensitive magnetic resonance imaging: a promising technique for depicting tissue characteristics containing neuromelanin. Neural Regen Res 2014, 9: 759.
Rozycka A, Jagodzinski P, Kozubski W, Lianeri M, Dorszewska J. Homocysteine level and mechanisms of injury in Parkinson’s disease as related to MTHFR, MTR, and MTHFD1 genes polymorphisms and L-Dopa treatment. Curr Genomics 2013, 14: 534–542.
Doherty GH. Homocysteine and Parkinson’s disease: a complex relationship. J Neurol Disord 2013, 1: 107.
Desforges NM, Hebron ML, Algarzae NK, Lonskaya I, Moussa CE. Fractalkine mediates communication between pathogenic proteins and microglia: implications of anti-inflammatory treatments in different stages of neurodegenerative diseases. Int J Alzheimers Dis 2012, 2012: 345472.
Wennström M, Surova Y, Hall S, Nilsson C, Minthon L, Boström F, et al. Low CSF levels of both α-synuclein and the α-synuclein cleaving enzyme neurosin in patients with synucleinopathy. PLoS One 2013, 8: e53250
Barone P et al. PRIAMO study group. The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov Disord 2009, 24: 1641–1649
Sharma S, Moon CS, Khogali A, Haidous A, Chabenne A, Ojo C, Jelebinkov M, Kurdi Y, Ebadi M. Biomarkers in Parkinson’s disease (recent update). Neurochem Int 2013, 63: 201–229.
Fekete R, Jankovic J. Revisiting the relationship between essential tremor and Parkinson’s disease. Mov Disord 2011, 26: 391–398.
Aarsland D, Brønnick K, Alves G, Tysnes OB, Pedersen KF, Ehrt U, et al. The spectrum of neuropsychiatric symptoms in patients with early untreated Parkinson’s disease. J Neurol Neurosurg Psychiatry 2009, 80: 928–930
Monderer R, Thorpy M. Sleep disorders and daytime sleepiness in Parkinson’s disease. Curr Neurol Neurosci Rep 2009, 9: 173–180
Ross GW, Petrovitch H, Abbott RD, Tanner CM, Popper J, Masaki K, et al. Association of olfactory dysfunction with risk for future Parkinson’s disease. Ann Neurol 2008, 63 : 167–173
Goldstein DS, Holmes C, Bentho O, Sato T, Moak J, Sharabi Y, et al. Biomarkers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Parkinsonism Relat Disord 2008, 14: 600–607.
Michell AW, Xu Z, Fritz D, Lewis SJ, Foltynie T, Williams-Gray CH, et al. Saccadic latency distributions in Parkinson’s disease and the effects of L-dopa. Exp Brain Res 2006, 174: 7–18.
Blair HA, Dhillon S. Safinamide: A review in Parkinson’s disease. CNS Drugs 2017, 31: 169–176.
Wang CD, Chan P. Clinicogenetics of Parkinson’s disease: a drawing but not completed picture. Neuroimmunol Neuroinflammation 2014, 1: 115.
Siddiqui IJ, Pervaiz N, Abbasi AA. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci Rep 2016, 6.
Oczkowska A, Kozubski W, Lianeri M, Dorszewska J. Mutations in PRKN and SNCA genes important for the progress of Parkinson’s disease. Curr Genomics 2013, 14: 502–517.
Kobo H, Bar-Shira A, Dahary D, Gan-Or Z, Mirelman A, Goldstein O, et al. Down-regulation of B cell-related genes in peripheral blood leukocytes of Parkinson’s disease patients with and without GBA mutations. Mol Genet Metab 2016, 117: 179–185.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lotankar, S., Prabhavalkar, K.S. & Bhatt, L.K. Biomarkers for Parkinson’s Disease: Recent Advancement. Neurosci. Bull. 33, 585–597 (2017). https://doi.org/10.1007/s12264-017-0183-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-017-0183-5