
Abstract
Tetanic stimulation of the sciatic nerve (TSS) triggers long-term potentiation in the dorsal horn of the spinal cord and long-lasting pain hypersensitivity. CX3CL1–CX3CR1 signaling is an important pathway in neuronal–microglial activation. Nuclear factor κB (NF-κB) is a key signal transduction molecule that regulates neuroinflammation and neuropathic pain. Here, we set out to determine whether and how NF-κB and CX3CR1 are involved in the mechanism underlying the pathological changes induced by TSS. After unilateral TSS, significant bilateral mechanical allodynia was induced, as assessed by the von Frey test. The expression of phosphorylated NF-κB (pNF-κB) and CX3CR1 was significantly up-regulated in the bilateral dorsal horn. Immunofluorescence staining demonstrated that pNF-κB and NeuN co-existed, implying that the NF-κB pathway is predominantly activated in neurons following TSS. Administration of either the NF-κB inhibitor ammonium pyrrolidine dithiocarbamate or a CX3CR1-neutralizing antibody blocked the development and maintenance of neuropathic pain. In addition, blockade of NF-κB down-regulated the expression of CX3CL1–CX3CR1 signaling, and conversely the CX3CR1-neutralizing antibody also down-regulated pNF-κB. These findings suggest an involvement of NF-κB and the CX3CR1 signaling network in the development and maintenance of TSS-induced mechanical allodynia. Our work suggests the potential clinical application of NF-κB inhibitors or CX3CR1-neutralizing antibodies in treating pathological pain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Many reports have confirmed that tetanic stimulation of the sciatic nerve (TSS) effectively induces long-term potentiation (LTP) of C- and A-fiber-evoked field potentials in the spinal dorsal horn, as well as long-lasting hypersensitivity to pain such as mechanical allodynia and thermal hyperalgesia [1,2,3]. LTP could also be regarded as a substrate for central sensitization of the pain pathway. But the mechanisms underlying the behavioral hypersensitivity caused by TSS still require further elucidation. Previously, we reported the cooperative activation of astrocytes and microglia during TSS-induced LTP [4]. On the other hand, we have also shown that P2X7 receptors might participate in the communication between microglia and neurons in the process of LTP evoked by TSS [3]. However, the intracellular (especially intranuclear) signaling that mediates the interaction between nerve cells remains largely unclear. Given that gliocytes participate in pathological pain, it is reasonable to hypothesize that inflammatory signals may be involved in the intracellular changes, so the transcription factor nuclear factor-kappa B (NF-κB), a frequently activated nuclear pathway in inflammatory cascades in neurons, may play a role in this induction. NF-κB, a key signal transduction molecule that regulates immuno-genes and inflammatory genes, is usually activated in the early phase of neuropathic pain [5]. In addition, CX3CL1 (fractalkine) and its receptor CX3CR1 are also known to regulate neuroinflammation in neuropathic pain via neuronal-glial signaling in the spinal cord [6,7,8,9,10]. Thus, we designed experiments to investigate whether TSS triggers NF-κB activation and alters the CX3CR1 expression in the dorsal horn and whether they participate in the TSS-induced development and maintenance of mechanical allodynia. Further, crosstalk between the NF-κB and CX3CL1/CX3CR1 axis was also probed. Our findings not only showed that these two pathways and their interaction are important participators in TSS-induced mechanical allodynia, but also provided interesting evidence for the spinal mechanism of mirror pain.
Materials and Methods
Animals
Sprague-Dawley rats (280–320 g; Shanghai Laboratory Animal Center, Shanghai Institutes for Biological Sciences) were group-housed (4 per cage) on a 12-h light/dark cycle at 18–23 °C, with free access to food and water. All animal experiments were approved by the Committee on the Use of Animal Experiments of Fudan University (Permit Number: SYXK 2009-0082) and followed the policies on the use of laboratory animals issued by the International Association for the Study of Pain.
Tetanic Stimulation of Sciatic Nerve (TSS)
Under pentobarbital sodium anesthesia (80 mg/kg, i.p.), the left sciatic nerve was carefully exposed at mid-thigh level and hung on a pair of silver hooks (only the sites touching the sciatic nerve were electrically conductive). The tetanic stimulation consisted of 10 trains of 0.5 ms rectangular pulses at 100 Hz and 40 V, 2 s in duration at 10-s intervals. After stimulation, the muscle and skin were sutured in layers. The sham-operated group received the same manipulation but without stimulation.
Von Frey Test for Mechanical Allodynia
Animals were allowed to acclimate for 30 min before testing. Each rat was placed in a chamber (20 × 10 × 20 cm3) on a platform with 10-mm grids of iron wires throughout the entire area. A series of von Frey filaments was applied to the central region of the plantar surface of one hind paw in ascending order (1 g, 1.4 g, 2 g, 4 g, 6 g, 8 g, 10 g, 15 g, and 26 g). Each filament was tested 5 times at 15-s intervals. The paw withdrawal threshold (PWT) was defined as the lowest force in grams that produced at least 4 withdrawal responses in 5 consecutive applications.
Drug Administration
Drugs were administered by lumbar puncture injection. Under isoflurane anesthesia, each rat was placed in a Plexiglas tube to widen the intervertebral spaces. No more than 15 μL of drug was delivered into the spinal space with a 30-gauge needle between the L5 and L6 vertebrae. PDTC (ammonium pyrrolidinedithiocarbamate, Sigma-Aldrich, St. Louis, MO; 100 ng/15 μL), rabbit anti-CX3CR1 (Torrey Pines Biolabs, Secaucus, NJ) or normal rabbit IgG (R&D Systems, Minneapolis, MN) was injected over a period of 4 min. Sterile normal saline was used as the solvent control.
Immunohistochemistry
Under deep anesthesia with an overdose of chloral hydrate (500 mg/kg, i.p.), animals were perfused intracardially with saline followed by 4% paraformaldehyde in 0.1 mol/L phosphate buffer (PBS; pH 7.4). The L4/L5 spinal segments were removed and postfixed overnight in 4% paraformaldehyde, which was then replaced with 20%–30% gradient sucrose in 0.1 mol/L PBS for 24–48 h at 4 °C. Tissues were frozen after OCT (optimal cutting temperature compound) embedding, and then cut at 30 μm on a freezing microtome (Leica, Wetzlar, Germany). For immunofluorescence staining, sections were washed, blocked with 10% donkey serum in 0.01 mol/L PBS with 0.3% Triton X-100 for 2 h, and incubated overnight at 4 °C with primary antibodies. On the next day, the sections were washed with PBS and incubated with secondary antibodies for 2 h, and then washed again. Sections were finally observed under a confocal laser-scanning microscope (FV1000, Olympus, Tokyo, Japan). The following primary antibodies were used: rabbit-anti-pNF-κB (1:200, Acris Antibodies GmbH, Herford, Germany), rabbit-anti-CX3CR1 (1:1000, Torrey Pines Biolabs, Secaucus, NJ), mouse anti-NeuN (1:1000, Millipore, Temecula, CA), mouse anti-GFAP (1:1000, Sigma-Aldrich, St Louis, MO), and mouse anti-OX-42 (1:1000, Bio-Rad, Oxford, UK, formerly Serotec). The secondary antibodies were RRX-labeled donkey anti-mouse or donkey anti-rabbit IgG (1:200, Jackson Immunoresearch, West Grove, PA) and FITC-labeled donkey anti-rabbit or donkey anti-mouse IgG (1:200, Jackson Immunoresearch, West Grove, PA). The specificity of immunostaining was verified by omitting the primary antibodies. The specificity of the primary antibodies was verified in pre-absorption experiments. Sections were first incubated with a mixture of primary antibody and the corresponding blocking peptide for 24 h, followed by secondary antibody incubation. The immunostaining signal was abolished after absorption.
Western Blots
After sacrifice, the L4–L6 spinal cord was rapidly removed. The spinal segments were cut into left and right halves along the ventral midline, and the left half was further split into dorsal and ventral horns at the level of the central canal. The dorsal horn tissues were homogenized and the protein concentrations measured. Samples of equal amounts of protein were loaded and separated on 10% SDS–PAGE gel and transferred to PVDF membranes. The membranes were blocked with 5% nonfat milk in Tris-buffered saline (pH 7.5) with 0.1% Tween-20 for 2 h at room temperature, and incubated overnight at 4 °C with primary antibodies. The blots were then incubated with HRP-conjugated secondary antibodies (1:1000, Thermo Scientific Pierce, Thermo Fisher Scientific Inc., Waltham, MA) for 2 h. Signals were detected using enhanced chemiluminescence (ECL, Thermo Scientific Pierce, Thermo Fisher Scientific Inc.), and the bands were analyzed with the ChemiDoc XRS system (Bio-Rad, Oxford, UK). The primary antibodies were rabbit anti-CX3CR1 (1:2000, Torrey Pines Biolabs, Secaucus, NJ) and rabbit-anti-pNF-κB (1:2000, Acris Antibodies GmbH, Herford, Germany).
ELISA Assay
A rat CX3CL1 “Sandwich” ELISA kit (RayBiotech, Norcross, GA) was used to assess CX3CL1 content. Rat recombinant fractalkine standards and samples in 100 μL were run in duplicate according to the manufacturer’s instructions. The optical density of each well was read at 450 nm.
Statistical Analysis
All data in this study are expressed as mean ± SEM. One-way or two-way repeated measures analysis of variance (RM-ANOVA) followed by the appropriate post hoc analysis was used to determine individual group differences. P < 0.05 was considered statistically significant.
Results
TSS Induces Activation of NF-κB in the Spinal Dorsal Horn
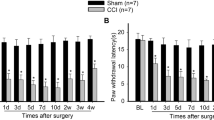
Consistent with our previous studies [11, 12], TSS induced a robust bilateral mechanical allodynia in rats. The PWTs for von Frey filaments were significantly decreased in both hind paws from day 1 after TSS and lasted for at least 10 days. The PWTs in the sham group did not show any significant changes on any day after surgery (Fig. 1A, B). Two-way RM-ANOVA revealed a significant effect of TSS (ipsilateral: F 1,18 = 227.31, P < 0.01; contralateral: F 1,18 = 214.42, P < 0.01) and an interaction between TSS treatment and time (ipsilateral: F 6,108 = 48.27, P < 0.01; contralateral: F 6,108 = 64.75, P < 0.01).

TSS induced bilateral mechanical allodynia and activation of NFκB in the spinal dorsal horn. A, B Ipsilateral and contralateral paw withdrawal thresholds decreased after tetanic stimulation of the sciatic nerve (TSS). C, D TSS increased the phosphorylated NF-κB (pNF-κB) levels on both sides of the dorsal horn at 1 days, 3 days, and 7 days. **P < 0.01 TSS vs Sham.
Corresponding to the mirror-like mechanical allodynia, western blot analysis showed significant up-regulation of pNF-κB levels on both sides of the dorsal horn at 1 days, 3 days, and 7 days after TSS (Fig. 1C, D; one-way ANOVA, F 4,20 = 21.731, P < 0.01), suggesting that NF-κB may be involved in the onset and maintenance of TSS-induced mechanical allodynia.
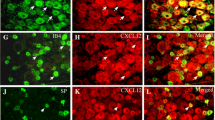
To determine the distribution of activated NF-κB in the dorsal horn, we performed double immunofluorescent staining for pNF-κB and NeuN (a neuronal marker), CD11 (a microglial marker, labeled by the antibody OX-42), or GFAP (an astrocytic marker) on sections of the L4–L6 spinal cord. Moderate pNF-κB immunoreactivity (IR) was found in all layers of the dorsal horn of sham-operated rats, and was exclusively co-localized with NeuN. TSS induced marked increases in the density and intensity of pNF-κB-IR cells in the ipsilateral spinal cord when examined 3 days after TSS, and a majority of pNF-κB-IR was found in NeuN-IR neurons, rarely in GFAP-IR astrocytes, and not in OX-42-IR microglia (Fig. 2).

Expression of pNF-κB in the ipsilateral dorsal horn on day 3 after TSS and sham-operation. The cell markers and pNF-κB were visualized by double immunofluorescent staining. NeuN, neuronal marker; GFAP, astrocytic marker; CD11 (labeled by the antibody OX-42), microglial marker. Scale bars, 25 μm.
NF-κB Inhibition Alleviates the Development and Maintenance of Mechanical Allodynia
To address the role of NF-κB in the development of TSS-induced mechanical allodynia, the NF-κB p65 inhibitor PDTC (100 ng/15 μL) was delivered intrathecally (i.t.) 1 h before and 12 h after TSS. TSS-induced mechanical allodynia was clearly alleviated by PDTC (Fig. 3A; two-way RM-ANOVA, treatment: F 1,12 = 41.26, P < 0.01; treatment × time: F 5,60 = 10.78, P < 0.01), while it did not affect the PWTs in sham-operated animals (Fig. 3B). To assess whether NF-κB is involved in the maintenance of TSS-induced pathological pain, the effects of PDTC on existing mechanical allodynia were examined 5 days after TSS. I.t. injection of PDTC (100 ng/15 μL) once daily for three consecutive days (days 5–7 after TSS) suppressed the established allodynia from day 7 to day 14 (Fig. 3C; two-way RM-ANOVA, treatment: F 1,12 = 9.839, P < 0.01; treatment × time: F 7,84 = 3.284, P < 0.01). Collectively, these findings suggest that NF-κB activation in the dorsal horn is necessary for both the development and maintenance of TSS-induced allodynia.

NF-κB inhibition alleviated the development of neuropathic pain and reversed existing mechanical allodynia. A PDTC alleviated the development of mechanical allodynia induced by TSS. B PDTC did not affect the PWTs in sham animals. C PDTC suppressed the established allodynia. *P < 0.05, **P < 0.01 PDTC vs NS.
CX3CR1 is Involved in TSS-Induced Mechanical Allodynia
Next, we assessed the changes of CX3CR1 expression after TSS. Similar to the activation of NF-κB, Western blot analysis showed that the expression of CX3CR1 was increased on both sides of the dorsal horn at all time points after TSS (Fig. 4A, B; one-way ANOVA, F 4,25 = 19.24, P < 0.01). Blockade of CX3CR1 by CX3CR1-neutralizing antibody (15 μg) at 1 h before and 12 h after TSS prevented the development of TSS-induced allodynia (Fig. 5A; two-way RM-ANOVA, treatment: F 1,12 = 33.84, P < 0.01; treatment × time: F 5,60 = 3.019, P = 0.016). I.t. injection of CX3CR1-neutralizing antibody or IgG did not affect the PWTs in sham-operated animals (Fig. 5B). Furthermore, when the mechanical allodynia stabilized at 5 days after TSS, the CX3CR1-neutralizing antibody reversed the existing allodynia (Fig. 5C; two-way RM-ANOVA, treatment: F 1,12 = 16.88, P < 0.01; treatment × time: F 5,60 = 3.017, P < 0.01).

CX3CR1 increased on both sides of the dorsal horn after TSS. The ipsilateral (A) and contralateral (B) CX3CR1 expression were detected by Western blot. **P < 0.01 TSS vs Sham.

Blockade of CX3CR1 alleviated the development of neuropathic pain and reversed existing mechanical allodynia. A CX3CR1 antibody prevented the development of TSS-induced allodynia. B CX3CR1 antibody did not affect the PWTs in sham-operated animals. C CX3CR1 antibody reversed the existing allodynia. **P < 0.01 CX3CR1 antibody vs IgG.
Interaction of NF-κB and CX3CL1–CX3CR1 Signaling
NF-κB regulates the expression of many cytokines and chemokines. To determine whether NF-κB signaling participates in the TSS-induced CX3CR1 upregulation, PDTC (100 ng/15 μL) was delivered i.t. 1 h before and 12 h after TSS, and the CX3CR1 expression levels were examined on days 1 and 3. The results showed that PDTC significantly downregulated the level of CX3CR1 on both day 1 and day 3 after TSS (Fig. 6A, B; one-way ANOVA, day 1: F 3,20 = 20.90, P < 0.01; day 3: F 3,20 = 74.49, P < 0.01). Moreover, we examined the effect of PDTC on the spinal CX3CL1 level. ELISA assays revealed that i.t. injection of PDTC (100 ng/15 μL, 1 h before and 12 h after TSS) significantly attenuated the TSS-induced CX3CL1 upregulation (Fig. 6C; one-way ANOVA: F 2,12 = 4.93, P = 0.027). Interestingly, i.t. injection of CX3CR1-antibody also inhibited the TSS-induced phosphorylation of NF-κB in the dorsal horn (Fig. 6D–F, one-way ANOVA, day 1: F 3,20 = 42.34, P < 0.01; day 3: F 3,20 = 78.38, P < 0.01). Together, these results highlighted possible cross-talk between neurons/astrocytes and microglia via NF-κB and CX3CL1/CX3CR1 signaling during the development and maintenance of pathological pain evoked by TSS.

NF-κB inhibition down-regulated CX3CR1 expression, and CX3CR1 blockade down-regulated the pNF-κB level in the dorsal horn. A, B PDTC down-regulated the CX3CR1 level on days 1 and 3 after TSS. C PDTC attenuated TSS-induced CX3CL1 expression on day 1. D–F CX3CR1 antibody inhibited the TSS-induced phosphorylation of NF-κB. *P < 0.05, **P < 0.01.
Discussion
The present study revealed an involvement of NF-κB and the CX3CL1/CX3CR1 axis in TSS-induced pathological pain. In particular, modulation of NF-κB or CX3CR1 signaling not only blocked the development and maintenance of mechanical allodynia but also inhibited the protein expression of each. This implied positive feedback between neurons/astrocytes and microglia during pathological pain.
The role of NF-κB in inflammation has been widely studied—it has been shown to modulate the expression of pro-inflammatory factors including IL-1β, IL-6, TNF-α, IL-17, and IL-18 [13,14,15,16], and chemokines such as CCL2, CXCL10, and CX3CL1 [17,18,19]. Activation of spinal NF-κB has been reported in pain models such as chronic constriction injury (CCI) [20, 21], diabetic neuropathic pain [22], and brachial plexus avulsion [23]. We further showed that TSS significantly increased the spinal pNF-κB level for >7 days. Inhibition of spinal NF-κB by i.t. PDTC before the development of allodynia robustly delayed or prevented TSS-induced mechanical allodynia. The allodynia established after TSS was also effectively relieved by PDTC. TSS has been used as a model of nerve injury [11]. Thus, the present results suggest the involvement of NF-κB activation in both the development and maintenance of TSS-induced pathological pain. Consistent with previous studies showing both pNF-κB and CX3CR1 upregulation in rats with CCI [24] and i.t. PDTC inhibition of CCI-induced CX3CR1 expression [25], our current results showed that following TSS, spinal CX3CR1 expression increased in parallel with NF-κB, and this was suppressed by the NF-κB inhibitor PDTC. Also, blockade of CX3CR1 prevented or alleviated TSS-induced allodynia. However, as shown above, pNF-κB was predominantly expressed in spinal neurons, in a few astrocytes, and not in microglia, whereas CX3CR1 in the dorsal horn is exclusively expressed in microglia in naive and in TSS-treated rats [26] (another report from our lab). A recent work from Xin’s laboratory showed that paclitaxel increases the recruitment of NF-κB p65 to the Cx3cl1 promoter region [19]. In support of this, we found that i.t. injection of PDTC significantly attenuated TSS-induced CX3CL1 in the dorsal horn. Also, our previous study revealed that spinal CX3CL1 occurs mostly in neurons and at a low level in astrocytes [26], suggesting a cellular distribution similar to NF-κB. Thus, we reasoned that inhibition of NF-κB might indirectly attenuate CX3CR1 via downregulating CX3CL1. In support, it has been reported that knockdown of CX3CL1 directly affects the expression of CX3CR1 [27]. Also, activation of NF-κB continually promotes the CX3CL1–CX3CR1 interaction [28]. In addition, other interactive networks may exist. For example, NF-κB could regulate IL-23 expression via NF-κB binding sites in the p19 subunit gene promoter of IL-23 [29]. PDTC has also been reported to reduce COX-2 [30] and the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α [31]. COX-2, IL-1β, IL-6, IL-23, and TNF-α have all been implicated in neuropathic pain, and may modulate the CX3CL1/CX3CR1 axis. Interestingly, here we not only demonstrated that NF-κB regulated CX3CR1 expression, but also, conversely, that blocking CX3CR1 decreased the pNF-κB level. In many cell systems, the CX3CL1/CX3CR1 axis has been shown to regulate the NF-κB pathway [32]. Ding et al. [33] further reported that, in mice with LPS-induced acute lung injury, activation of the CX3CL1/CX3CR1 axis increases the level of NF-κB phosphorylation, and CX3CL1-knockout mice exhibit decreased pNF-κB level. There are still some limitations of our study on the crosstalk between NF-κB and CX3CR1. Although blockade of CX3CR1 inhibited TSS-induced pNF-κB, we do not know which molecules and signals are involved. Further study is needed to explore the direct target molecule for CX3XR1 and NF-κB regulation.
Another interesting outcome of the present study was that unilateral TSS induced bilateral mechanical allodynia and bilateral upregulation of pNF-κB and CX3CR1. A similar phenomenon has been reported in other independent mirror-pain studies. Choi et al. [34] reported that spinal IL-1β and astrocytic connexin 43 play a role in the development of mirror-image pain, but intriguingly, they also pointed out that IL-1β is expressed by microglia and capable of suppressing astrocyte activation, ultimately preventing the development of contralateral mirror pain [35]. Although the mechanism of mirror-image pain has not been fully determined, a general consensus is that some mediators (inflammatory mediators in particular) spread to the contralateral spinal cord and activate the corresponding effector cells [36,37,38]. This study is among the very few suggesting that microglia are involved in the mechanism underlying mirror pain. Different from those of Choi, our findings support the hypothesis that microglia play a contributory rather than a suppressive role. In any case, further research is needed for more compelling evidence.
In summary, in the present study we showed that unilateral TSS induced bilateral mechanical allodynia and up-regulated the expression of phosphorylated NF-κB and CX3CR1. Blockade of NF-κB and CX3CR1 not only attenuated/reversed the TSS-induced allodynia, but also down-regulated the expression of each. Thus, NF-κB and the CX3CR1 network may be important targets for new approaches to pain relief.
References
Liu X, Sandkuhler J. Characterization of long-term potentiation of C-fiber-evoked potentials in spinal dorsal horn of adult rat: essential role of NK1 and NK2 receptors. J Neurophysiol 1997, 78: 1973–1982.
Zhang XC, Zhang YQ, Zhao ZQ. Different roles of two nitric oxide activated pathways in spinal long-term potentiation of C-fiber-evoked field potentials. Neuropharmacology 2006, 50: 748–754.
Chu YX, Zhang Y, Zhang YQ, Zhao ZQ. Involvement of microglial P2X7 receptors and downstream signaling pathways in long-term potentiation of spinal nociceptive responses. Brain Behav Immun 2010, 24: 1176–1189.
Chu YX, Zhang YQ, Zhao ZQ. Involvement of microglia and interleukin-18 in the induction of long-term potentiation of spinal nociceptive responses induced by tetanic sciatic stimulation. Neurosci Bull 2012, 28: 49–60.
Ledeboer A, Gamanos M, Lai W, Martin D, Maier SF, Watkins LR, et al. Involvement of spinal cord nuclear factor kappaB activation in rat models of proinflammatory cytokine-mediated pain facilitation. Eur J Neurosci 2005, 22: 1977–1986.
Harrison J K, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, et al. Role for neuronal derived fratalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A 1998, 95: 10896–10901.
Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci 2004, 20: 1150–1160.
Sun S, Cao H, Han M, Li TT, Pan HL, Zhao ZQ, et al. New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glial activation in rat model of monmarthritis. Pain 2007, 129: 64–75.
Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, et al. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci 2011, 31: 9910–9922.
Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun 2007, 21:642–651.
Liang L, Wang Z, Lv N, Yang J, Zhang Y, Zhao Z. Involvement of nerve injury and activation of peripheral glial cells in tetanic sciatic stimulation-induced persistent pain in rats. J Neurosci Res 2010, 88: 2899–2910.
Liang LL, Yang JL, Lü N, Gu XY, Zhang YQ, Zhao ZQ. Synergetic analgesia of propentofylline and electroacupuncture by interrupting spinal glial function in rats. Neurochem Res 2010, 35: 1780–1786.
Tolosa L, Caraballo-Miralles V, Olmos G, Llado J. TNF-alpha potentiates glutamate-induced spinal cord motoneuron death via NF-kappaB. Mol Cell Neurosci 2011, 46: 176–186.
Zong S, Li K, Zeng G, Fang Y, Zhao J. The Effects of interleukin-17 (IL-17)-related inflammatory cytokines and A20 regulatory proteins on astrocytes in spinal cord cultured in vitro. Cell Physiol Biochem 2016, 38: 1100–1110.
Kim J, Shao Y, Kim SY, Kim S, Song HK, Jeon JH, et al. Hypoxia-induced IL-18 increases hypoxia-inducible factor-1alpha expression through a Rac1-dependent NF-kappaB pathway. Mol Biol Cell 2008, 19: 433–444.
Guzman-Soto I, Salinas E, Quintanar JL. Leuprolide acetate inhibits spinal cord inflammatory response in experimental autoimmune encephalomyelitis by suppressing NF-kappaB activation. Neuroimmunomodulation 2016, 23: 33–40.
Kaltschmidt B, Kaltschmidt C. NF-kappaB in the nervous system. Cold Spring Harb Perspect Biol 2009, 1: a001271.
Richmond A. NF-kappa B, chemokine gene transcription and tumour growth. Nat Rev Immunol 2002, 2: 664–674.
Li D, Huang ZZ, Ling YZ, Wei JY, Cui Y, Zhang XZ, et al. Up-regulation of CX3CL1 via nuclear factor-κb-dependent histone acetylation is involved in paclitaxel-induced peripheral neuropathy. Anesthesiology 2015, 122:1142–151.
Gui Y, Li A, Chen F, Zhou H, Tang Y, Chen L, et al. Involvement of AMPK/SIRT1 pathway in anti-allodynic effect of troxerutin in CCI-induced neuropathic pain. Eur J Pharmacol 2015, 769: 234–241.
Chen Y, Chen X, Yu J, Xu X, Wei X, Gu X, et al. JAB1 is involved in neuropathic pain by regulating JNK and NF-kappaB activation after chronic constriction injury. Neurochem Res 2016, 41: 1119–1129.
Zhang YP, Song CY, Yuan Y, Eber A, Rodriguez Y, Levitt RC, et al. Diabetic neuropathic pain development in type 2 diabetic mouse model and the prophylactic and therapeutic effects of coenzyme Q10. Neurobiol Dis 2013, 58: 169–178.
Paszcuk AF, Dutra RC, da Silva KA, Quintao NL, Campos MM, Calixto JB. Cannabinoid agonists inhibit neuropathic pain induced by brachial plexus avulsion in mice by affecting glial cells and MAP kinases. PLoS One 2011, 6: e24034.
Cao H, Zheng JW, Li JJ, Meng B, Li J, Ge RS. Effects of curcumin on pain threshold and on the expression of nuclear factor κ B and CX3C receptor 1 after sciatic nerve chronic constrictive injury in rats. Chin J Integr Med 2014, 20: 850–856.
Pan YD, Guo QL, Wang E, Ye Z, He ZH, Zou WY, et al. Intrathecal infusion of pyrrolidine dithiocarbamate for the prevention and reversal of neuropathic pain in rats using a sciatic chronic constriction injury model. Reg Anesth Pain Med 2010, 35:231–237.
Bian C, Wang ZC, Yang JL, Lu N, Zhao ZQ, Zhang YQ. Up-regulation of interleukin-23 induces persistent allodynia via CX3CL1 and interleukin-18 signaling in the rat spinal cord after tetanic sciatic stimulation. Brain Behav Immun 2014, 37: 220–230.
Jamieson WL, Shimizu S, D’Ambrosio JA, Meucci O, Fatatis A. CX3CR1 is expressed by prostate epithelial cells and androgens regulate the levels of CX3CL1/fractalkine in the bone marrow: potential role in prostate cancer bone tropism. Cancer Res 2008, 68: 1715–1722.
Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, et al. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett 1998, 429: 167–172.
Mise-Omata S, Kuroda E, Niikura J, Yamashita U, Obata Y, Doi TS. A proximal kappaB site in the IL-23 p19 promoter is responsible for RelA- and c-Rel-dependent transcription. J Immunol 2007, 179: 6596–6603.
Lavon I, Goldberg I, Amit S, Landsman L, Jung S, Tsuberi BZ, et al. High susceptibility to bacterial infection, but no liver dysfunction, in mice compromised for hepatocyte NF-kappaB activation. Nat Med 2000, 6: 573–577.
Li J, Zhao PP, Hao T, Wang D, Wang Y, Zhu YZ, et al. Urotensin II inhibitor eases neuropathic pain by suppressing the JNK/NF-κB pathway. J Endocrinol 2017, 232: 165–174.
McComb JG, Ranganathan M, Liu XH, Pilewski JM, Ray P, Watkins SC, et al. CX3CL1 up-regulation is associated with recruitment of CX3CR1+ mononuclear phagocytes and T lymphocytes in the lungs during cigarette smoke-induced emphysema. Am J Pathol 2008, 173: 949–961.
Ding XM, Pan L, Wang Y, Xu QZ. Baicalin exerts protective effects against lipopolysaccharide-induced actue lung injury by regulating the crosstalk between the CX3CL1-CX3CR1 axis and NF-kB pathway in CX3CL1-knockout mice. Int J Mol Med 2016, 37: 703–715.
Choi HS, Roh DH, Yoon SY, Kwon SG, Choi SR, Kang SY, et al. The role of spinal interleukin-1beta and astrocyte connexin 43 in the development of mirror-image pain in an inflammatory pain model. Exp Neurol 2016, 287: 1–13.
Choi HS, Roh DH, Yoon SY, Moon JY, Choi SR, Kwon SG, et al. Microglial interleukin-1beta in the ipsilateral dorsal horn inhibits the development of mirror-image contralateral mechanical allodynia through astrocyte activation in a rat model of inflammatory pain. Pain 2015, 156: 1046–1059.
Lv H, Chen H, Xu JJ, Jiang YS, Shen YJ, Zhou SZ, et al. Redox imbalance in the peripheral mechanism underlying the mirror-image neuropathic pain due to chronic compression of dorsal root ganglion. Neurochem Res 2016, 41: 958–964.
Cheng CF, Cheng JK, Chen CY, Rau RH, Chang YC, Tsaur ML. Nerve growth factor-induced synapse-like structures in contralateral sensory ganglia contribute to chronic mirror-image pain. Pain 2015, 156: 2295–2309.
Cheng CF, Cheng JK, Chen CY, Lien CC, Chu D, Wang SY, et al. Mirror-image pain is mediated by nerve growth factor produced from tumor necrosis factor alpha-activated satellite glia after peripheral nerve injury. Pain 2014, 155: 906–920.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81471130, 31420103903, and 31421091) and a Development Project of Shanghai Peak Disciplines Integrated Chinese and Western Medicine.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Wang, ZC., Li, LH., Bian, C. et al. Involvement of NF-κB and the CX3CR1 Signaling Network in Mechanical Allodynia Induced by Tetanic Sciatic Stimulation. Neurosci. Bull. 34, 64–73 (2018). https://doi.org/10.1007/s12264-017-0149-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-017-0149-7