
Abstract
The application of neurotrophic factors (NTFs) is a promising therapeutic strategy for neurodegenerative disorders such as Parkinson’s disease (PD). Many NTFs have been reported to enhance the survival, regeneration, and differentiation of neurons and to induce synaptic plasticity. However, because of their potential side-effects and low efficacy after clinical administration, more potent treatments for neurodegenerative disorders are being sought. Cerebral dopamine neurotrophic factor (CDNF), a newly-identified NTF homologous to mesencephalic astrocyte-derived NTF, is structurally and functionally different from other NTFs, providing new hope especially for PD patients. In various animal models of PD, CDNF is efficient in protecting and repairing dopaminergic neurons, and it inhibits endoplasmic reticulum stress, neuroinflammation, and apoptosis. Recent progress in all facets of CDNF research has enabled researchers to better understand its beneficial effects in the treatment of PD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neurotrophic factors (NTFs) are peptides essential for the growth, maturation, and survival of neurons and axons. Alterations in the levels and functions of these secreted proteins have been associated with various neurodegenerative disorders such as Parkinson’s disease (PD). To date, many NTFs have been discovered and identified as having diverse biological effects; they include the neurotrophins nerve growth factor, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and NT-4; glial cell-line derived neurotrophic factor (GDNF) and its family of ligands, such as neurturin; neurotrophic cytokines; and the recently-characterized cerebral dopamine NTF (CDNF) and the mesencephalic astrocyte-derived NTF (MANF) [1,2,3,4]. In recent years, several NTFs have been used to treat human diseases. Since they play substantial roles in controlling survival and death pathways in neurons, specific factors have received increasing attention for the treatment of neurodegenerative disorders [5].
The pathogenesis of PD is complex and involves several mechanisms, leading to a cohort of motor symptoms and cognitive impairments, as well as sensory, sleep, and gastrointestinal dysfunctions [6]. Currently there is no cure. The typical treatment aims to alleviate the symptoms rather than the synaptic loss and neuronal death. The new neurogenic agents, including NTFs, represent a novel strategy for addressing the degenerative process itself. Mounting evidence has shown that numerous NFTs act to protect dopamine (DA) neurons from selective degeneration [7, 8]. CDNF has been demonstrated to have potential therapeutic benefits for PD [3]. With respect to the pathogenesis of PD, CDNF acts as a robust endogenous agent that provides protection and recovery particularly of DA neurons in laboratory and clinical studies. In these contexts, CDNF has been used as an antioxidant, anti-inflammatory, and anti-apoptotic agent to improve mitochondrial function and prevent the cytotoxic aggregation of proteins [9, 10]. Furthermore, CDNF administration also relieves the lesions in the 6-hydroxydopamine (6-OHDA) rat and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse models [8, 11]. In this review, we summarize the basic insights and laboratory investigations regarding CDNF as a therapeutic agent for PD, as well as the underlying molecular mechanisms by which it protects and repairs DA neurons.
Structure and Distribution of CDNF
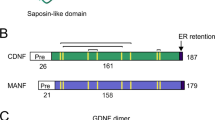
CDNF was first discovered by Lindholm using a bioinformatics approach, and then its conformation was characterized via biochemical methods [3]. Prior to that study, MANF was initially identified from a rat mesencephalic type 1 astrocyte cell line in 2003 [4]. As CDNF and MANF share common functional domains, studies of MANF are relevant to the role of CDNF. CDNF is a paralog of MANF, and human CDNF shows 59% amino-acid identity with human MANF [3]. Together, CDNF and MANF form a new, evolutionarily-conserved family of NTFs due to their unique structures and potent protection of embryonic DA neurons [12, 13]. These proteins are characterized by three-dimensional structures containing eight α-helices [14, 15], and they differ from all other known NTFs and growth factors in amino-acid sequence. The CDNF protein is an 18-kDa monomer consisting of 187 amino-acids (containing 26 amino-acids in its signal peptide), and its encoding gene contains 4 exons that are highly evolutionarily conserved [16]. Analysis of CDNF crystal structure indicated that two domains are essential for CDNF to fully exert its function. One is the N-terminus, which is a saposin-like structure, and the other is the C-terminus, which contains a disulfide bridge between two cysteine residues [12, 17]. The N-terminus has a lipid-binding capacity that allows the saposin-like structure to transfer across the lipid membranes. In comparison with the N-terminus, the C-terminus might have a marked effect through the unfolded Cys-Xaa-Xaa-Cys (CXXC) domain and a putative endoplasmic reticulum (ER) retention signal sequence (KTEL), indicating that CDNF might be involved in ER stress, which is an important component of PD pathogenesis [18,19,20,21]. Mutation of the CXXC motif of the C-terminal domain abolishes the actions of MANF and CDNF; in contrast, expression of the C-terminal domain rescues sympathetic neurons from toxin-induced apoptosis in a Drosophila model [22, 23]. In PD patients, the C allele of an intronic CDNF single nucleotide polymorphism (rs7094179) has been associated with susceptibility to PD [24]. In addition, human CDNF has two structural isoforms, the non-glycosylated [3] and the glycosylated conformations [17], but neither is found in rodent CDNF. Emerging evidence supports the notion that glycosylation of CDNF is not required for its secretion, but helix 7 is essential for its regulated secretion, and helix 1 is vital for both constitutive and regulated secretion [25].
In mammals, CDNF expression occurs broadly in the central nervous system and in peripheral tissues, including the embryonic, postnatal, and adult mouse brain as well as the adult human brain [26]. Transcript detection via RT-PCR and in situ hybridization has found CDNF transcripts in almost all brain regions, including the striatum, corpus callosum, and optic nerve [3], and its transcription can be regulated by the clinical drug valproic acid [27, 28]. Among peripheral organs, higher CDNF mRNA expression has been reported in the adult mouse heart, skeletal muscle, and testis than in the lung and stomach [3, 29], although the overall expression level is lower than that of MANF [29, 30]. Consistent with mRNA expression analysis, CDNF protein expression has been detected in various areas of the adult mouse brain, including the cerebral cortex, hippocampus, cerebellum, thalamus, substantia nigra (SN), and striatum, showing a partial overlap with MANF expression [3, 29], indicating that CDNF targets various cerebral territories to perform broad functions. One interesting finding is that the gene expression levels of CDNF as well as GDNF are down-regulated in the SN under conditions of space flight, presumably explaining the deleterious effects of microgravity on the DA system [31].
Secretion of CDNF
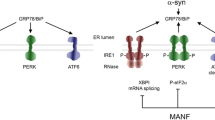
Abundant evidence has demonstrated that both MANF and CDNF are secreted, with fundamentally similar secretory mechanisms, following the classic ER-Golgi pathway. Further, their neuroprotective roles are induced by ER stress, and the secreted proteins can prevent cell death from such stress [17, 32, 33]. Although studies have shown that the endogenous protein expression and secretion of MANF are induced by ER stress in various cell types both in vivo and in vitro [17, 34], little is known about the secretion of CDNF during ER stress or physiological stimuli. In cultures of the cell lines U2OS and HEK293, the expression and secretion of CDNF are not regulated by activation of the unfolded protein response (UPR), a signaling pathway induced by various agonists [17]. In contrast to BDNF, which is secreted from vesicles via exocytosis [35], CDNF is not secreted via the intrinsic secretory pathway but rather via the regulated secretory pathway [25]. CDNF is commonly glycosylated in cells by both N-linked and O-linked glycosylation, but glycosylation has no effect on its secretion. Furthermore, two substructures in CDNF, the α-1 and α-7 helices, affect its intracellular storage and secretion. Inhibition of glycosylation of these substructures markedly reduces the secretion of CDNF and causes its retention in the ER, preventing its transfer into the Golgi complex for further processing [25].
Functions and Protective Mechanisms of CDNF in Disease Models
Like other NTFs, both CDNF and MANF are considered to be candidate therapeutic agents for the treatment of neurodegenerative diseases. Although MANF has been shown to be a secreted mediator of the adaptive pathway regulated by the UPR and to protect against cell death from ER stress, apoptosis, and inflammation [36, 37], the functional mechanisms of action of MANF and CDNF seem to be distinct. MANF enhances the survival of embryonic DA neurons in vitro and increases the γ-aminobutyric acid-A receptor-mediated inhibition of postsynaptic currents in DA neurons [4, 38], whereas CDNF is best known for its protection and repair of DA neurons in the SN. In vivo, CDNF prevents the 6-OHDA-induced degeneration of DA neurons in the rat model of PD. It appears to be as potent as GDNF in having a long-term beneficial effect to reduce amphetamine-induced rotational behavior [3]. CDNF not only alleviates 6-OHDA-induced TH-positive cell death in the SN but also inhibits the loss of TH-positive fibers in the striatum [39]. In the MPTP-induced mouse model of PD, CDNF also shows effective neuroprotection, increasing the number of DA neurons in the SN and improving horizontal and vertical motor behaviors [8]. Physiologically, CDNF has been shown to prevent the DA neuron damage caused by α-synuclein oligomers [10]. In addition to the potential for clinical therapy for PD patients, CDNF has another important function of improving memory. In the APP/PS1 transgenic mouse model of Alzheimer’s disease (AD), injection of CDNF protein or CDNF transgenic AAV2 viral vectors into the hippocampus of 1-year-old animals followed by the water-maze test has shown that CDNF-treated mice reach the platform more quickly and more accurately, and they stay longer on the platform, suggesting that CDNF expression in neurons is closely related to long-term memory. Unexpectedly, CDNF does not affect the spontaneous exploration or the early spatial learning of targets [40]. Moreover, pretreatment with CDNF reduces the expression levels of ER stress-related proteins and amyloid-beta-induced synaptotoxicity at an early stage of AD [41].
Peripheral nerve injury induces axonal degeneration and neuronal loss, thereby affecting sensory and motor functions [42, 43]. Accumulating evidence has demonstrated that lentiviral vectors containing the CDNF gene are effectively expressed in many models of nerve regeneration, providing insight into the role of CDNF in nerve regeneration and functional recovery [44,45,46]. Therefore, CDNF can be applied via lentiviral expression in animal models. However, the concentration is important for the application of CDNF; Garea-Rodríguez and colleagues showed that the long-term administration of high-dose CDNF leads to pathological changes in primate brain, and such doses, usually 7% of the maximum tolerated dose, have potential immunogenicity in individual animals [47]. Although CDNF is a promising protective and restorative factor in various animal models of neurological diseases, the mechanisms underlying these effects have received much less attention. Although CDNF has both anti-inflammatory and anti-apoptotic effects, current research is mainly focused on animal models of PD (Table 1).
Nerve Repair and Protection
As a common age-related neurodegenerative disorder, PD is characterized by a progressive loss of DA neurons and the accumulation of Lewy bodies in the SN [48, 49]. Thus, the actions of CDNF in nerve repair and protection may be significant for the treatment of PD.
CDNF has been found to prevent the degeneration of DA neurons in response to 6-OHDA neurotoxicity in rats. A single injection of CDNF before 6-OHDA lesioning or four weeks of CDNF administration after 6-OHDA lesioning in the striatum significantly reduces the amphetamine-induced rotation and rescues DA function in the SN [3]. Using a rat model, Voutilainen and co-workers showed that continuous infusion of CDNF for two weeks following an intrastriatal 6-OHDA lesion is able to alleviate the rotational behavior and also effectively inhibits the 6-OHDA-induced loss of TH-positive neurons in the SN and of TH-immunoreactive fibers in the striatum [39]. In normal animals, long-term injection of CDNF affects behavioral habits and the pathway from the striatum to the SN, and the route of CDNF transport from the striatum to the SN is reverse-selective transport, rather than multiple-point transport. This study therefore revealed an important step for CDNF to act as a therapeutic agent for PD [39, 50]. Experimentally, these effects of CDNF seem more promising than those of MANF and GDNF, likely attributable to the difference in diffusion volume, given that the distribution of CDNF is greater than that of GDNF in the striatum after long-term injection using an 125I-labeling strategy [39, 51]. However, the physiological differences between CDNF and MANF are unknown. Recently, a comparative analysis of the molecular features of mouse MANF and CDNF showed that their different physiological actions could be ascribed to the evolutionary differences in their C-terminal amino-acids [33].
In addition to the 6-OHDA-induced models, the protective and reparative effects of CDNF have also been characterized in the MPTP-induced mouse model of PD. A study revealed that transferring CD3-activated regulatory T cells to MPTP-intoxicated mice up-regulates CDNF expression [52]. Airavaara et al. constructed PD models by injecting MPTP into C57/BL6 mice to determine whether CDNF protects the DA system and induces neuron repair. They found that CDNF pretreatment increases TH-immunoreactivity in both the SN and striatum, as well as the number of DA neurons in the SN. Consistently, CDNF treatment after MPTP lesions alleviates the horizontal and vertical motor behavior of mice and restores DA fiber density [8]. In healthy rodents, GDNF is well known to promote axonal development and improve DA uptake in the SN [53, 54], while CDNF has no effect on the nigrostriatal DA system [8]. Recently, the therapeutic effect of CDNF has been compared with that of GDNF with regard to the protection of DA neurons in a unilateral 6-OHDA-lesioned marmoset model of PD. CDNF treatment significantly increased the DA transporter binding activity in lesioned animals, with a therapeutic efficacy more potent than that of GDNF [47].
To assess the action of CDNF in improving peripheral nerve regeneration, the effects of overexpressing CDNF protein have been studied in a rat model of sciatic nerve transection. The results showed that the axons had regenerated at four weeks and the Schwann cells eight weeks after injury in the CDNF-treated group, and the myelin around central axons in the CDNF-treated group was clearly thicker than that in the control group [46].
These different animal experiments have confirmed that CDNF functions in neuroprotection and regeneration, which provides an important theoretical basis for the clinical treatment of neurodegenerative diseases, particularly PD.
Anti-inflammatory Effects
In addition to the neuroprotective effects, accumulating evidence has shown that CDNF has an anti-inflammatory action. Importantly, CDNF relieves ER-stress-induced astrocyte injury and inhibits the secretion of pro-inflammatory factors [15]. The inflammatory reaction is a major pathogenic process of PD. Rocha and colleagues demonstrated that CDNF expression is present in primary astrocytes in the midbrain [55]. Overexpression of CDNF in primary astrocytes alleviates ER stress-induced cellular damage and suppresses the secretion of pro-inflammatory cytokines induced by tunicamycin [56]. In rodent models transiently transfected with the human CDNF gene, 6-OHDA triggers nitrosative stress in the SN, increases the levels of inflammatory cytokines, and activates neuron/glial type 2 cells to convert into astrocytes, leading to a high production of CDNF. Significantly, CDNF inhibits the 6-OHDA-induced neuroinflammation by decreasing the levels of nitrosative stress and IL-6 in the SN, thereby having an anti-inflammatory effect [9]. Furthermore, lipopolysaccharide (LPS) up-regulates CDNF in cultured rat primary microglia, and CDNF administration reduces the LPS-induced cytotoxicity and the production of pro-inflammatory cytokines via inhibiting the JNK signaling pathway [57]. These data provide evidence that CDNF acts as a neuroprotective agent by interfering with the neuroinflammatory response in the central nervous system.
Anti-apoptotic Functions
Apoptosis is an important indicator of the degree of neurological dysfunction in neurodegenerative diseases [58]. Although the cellular signals involved in apoptosis are not fully understood, activation of ER stress results in an enhanced sensitivity to apoptosis [59,60,61]. One hypothesis for how CDNF prevents the degeneration of DA neurons is that it suppresses apoptosis at least partially by inhibiting the ER stress, the mechanism of which has been reviewed elsewhere [62]. In vitro, 6-OHDA dramatically decreases the viability of PC12 cells. However, either pre-treatment or post-treatment with CDNF significantly enhances the cell survival by modulating the apoptotic pathway through increasing the Bcl-2/Bax ratio and downregulating the caspase-3 activity (Fig. 1), and these effects occur in a dose-dependent manner [63, 64]. Notably, the C-terminal domain of MANF is homologous to the SAP domain of Ku70, a well-known inhibitor of the pro-apoptotic protein Bax [13, 65]. Although the crystal structure of the C-terminal domain of CDNF is very similar to that of MANF [10], how CDNF blocks apoptosis via interacting with Bax-associated pathways is currently unknown.

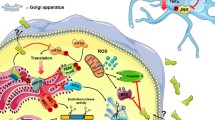
Schematic of the proposed neuroprotective mechanism of action of CDNF in dopaminergic neurons. In the PD model, CDNF binds to an unknown receptor that activates the intracellular signaling pathways and effectively inhibits 6-OHDA-induced ROS production, which in turn affects the Bcl-2/Bax ratio and caspase-3 apoptotic signals. Moreover, the endogenous CDNF is retained in the ER where it directly affects the ER stress and UPR pathways. In the ER-stressed cells, CDNF could inhibit apoptosis induced by three UPR sensors (PERK, IRE1, and ATF6). ATF6 activating transcription factor 6, ER endoplasmic reticulum, GRP78 glucose regulated protein 78, IRE1 inositol-requiring enzyme 1, PERK pancreatic ER kinase-like ER kinase, ROS reactive oxygen species, UPR unfolded protein response.
Cell-Based and Gene-Therapy Approaches
Like other NTFs, the clinical application of CDNF faces many challenges such as poor penetration of the blood-brain barrier. Repeated injection or infusion into the brain raises the risk of infection. However, cell-based transplantation and engineered gene-expression therapies have provided biological vehicles for the long-term administration of CDNF and produced promising outcomes for targeting the DA system. Primary cell cultures stably expressing CDNF can be derived from small-volume brain biopsies in PD patients during surgery for deep brain stimulation, suggesting the feasibility of small brain biopsies as an autologous cell source for therapeutic application in PD [66]. Recently, CDNF-expressing bone marrow stromal cells (BMSCs) or CDNF-expressing bone marrow-derived mesenchymal stem cells (MSCs) were injected into the striatum of rats with 6-OHDA-induced neurotoxicity, and they reduced DA neuron loss and restored the tyrosine hydroxylase levels and reduced the PD-related behavior [67, 68]. In addition to PD, transplantation of CDNF-BMSCs also suppresses neuroinflammation by reducing the production of pro-inflammatory cytokines after traumatic spinal cord injury [69]. Unfortunately, these studies do not provide evidence regarding how exogenous cell grafts escape immunological barriers and integrate into the host microenvironment.
Gene-therapy approaches using NTFs have been accepted in the clinical management of PD symptoms [51]. Recombinant adeno-associated viruses (AAVs) with minor adverse effects are widely used for gene delivery in neurodegenerative diseases [70, 71]. CDNF delivered into the striatum using a type 2 AAV restores the functions of DA neurons in the SN and fiber density in the striatum, while effectively suppressing the 6-OHDA-induced behavioral deficits in rats [72]. Consistent results were obtained in a study of rats undergoing a similar treatment; delivery of the CDNF gene into the striatum gave rise to long-lasting protein expression, resulting in the reduction of amphetamine-induced rotation [11]. Moreover, the effects of CDNF delivery using a bicistronic lentiviral vector have also been studied. CDNF expressed by a lentiviral vector inhibits dendrite formation in hypothalamic neurons in animal models of PD [73]. Cordero-Llana and co-workers reported that lentiviral vector-mediated overexpression of CDNF in the SN reduces 6-OHDA-induced behavioral abnormalities and restores the DA fiber density but does not change the number of DA neurons in the SN [74]. Importantly, that study also showed that nigral delivery of CDNF is more effective than striatal delivery, and the combined administration of CDNF and MANF has synergistic effects in the neuroprotection and repair of DA neurons [74].
Conclusions
In comparison with other NTFs, CDNF shows multiple advantages: (1) a lower affinity for the extracellular matrix, (2) a large range of diffusion, (3) a selective effect on DA neurons, (4) no influence on peripheral motor, sensory, and sympathetic neurons, (5) fewer side-effects, and (6) greater effects with low or medium concentrations than GDNF and BDNF. Although CDNF has certain beneficial effects on the adult midbrain DA system, validating the safety and rapid intra-parenchymal delivery of CDNF requires additional preclinical studies before its general adoption for the clinical treatment of PD and other neurodegenerative diseases.
Studies on animal models of PD have shown that CDNF has potential therapeutic uses; however, there is still a long way to go before we achieve a better performance of CDNF in clinical trials. To date, there is little information about the basic biology and corresponding plasma membrane receptors of CDNF, and its effects on other central or peripheral neurons remain unclear. Remarkably, orchestration of the intracellular signaling pathways triggered by CDNF might offer an alternative strategy for the use of neurotrophic factors in clinical therapies.
References
Deister C, Schmidt CE. Optimizing neurotrophic factor combinations for neurite outgrowth. J Neural Eng 2006, 3: 172–179.
Rodrigues TM, Jeronimo-Santos A, Outeiro TF, Sebastiao AM, Diogenes MJ. Challenges and promises in the development of neurotrophic factor-based therapies for Parkinson’s disease. Drugs Aging 2014, 31: 239–261.
Lindholm P, Voutilainen MH, Lauren J, Peranen J, Leppanen VM, Andressoo JO, et al. Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 2007, 448: 73–77.
Petrova P, Raibekas A, Pevsner J, Vigo N, Anafi M, Moore MK, et al. MANF: a new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J Mol Neurosci 2003, 20: 173–188.
Rangasamy SB, Soderstrom K, Bakay RA, Kordower JH. Neurotrophic factor therapy for Parkinson’s disease. Prog Brain Res 2010, 184: 237–264.
Batla A, Tayim N, Pakzad M, Panicker JN. Treatment options for urogenital dysfunction in Parkinson’s disease. Curr Treat Options Neurol 2016, 18: 45.
Su YR, Wang J, Wu JJ, Chen Y, Jiang YP. Overexpression of lentivirus-mediated glial cell line-derived neurotrophic factor in bone marrow stromal cells and its neuroprotection for the PC12 cells damaged by lactacystin. Neurosci Bull 2007, 23: 67–74.
Airavaara M, Harvey BK, Voutilainen MH, Shen H, Chou J, Lindholm P, et al. CDNF protects the nigrostriatal dopamine system and promotes recovery after MPTP treatment in mice. Cell Transplant 2012, 21: 1213–1223.
Nadella R, Voutilainen MH, Saarma M, Gonzalez-Barrios JA, Leon-Chavez BA, Jimenez JM, et al. Transient transfection of human CDNF gene reduces the 6-hydroxydopamine-induced neuroinflammation in the rat substantia nigra. J Neuroinflammation 2014, 11: 209.
Latge C, Cabral KM, de Oliveira GA, Raymundo DP, Freitas JA, Johanson L, et al. The solution structure and dynamics of full-length human cerebral dopamine neurotrophic factor and its neuroprotective role against alpha-synuclein oligomers. J Biol Chem 2015, 290: 20527–20540.
Back S, Peranen J, Galli E, Pulkkila P, Lonka-Nevalaita L, Tamminen T, et al. Gene therapy with AAV2-CDNF provides functional benefits in a rat model of Parkinson’s disease. Brain Behav 2013, 3: 75–88.
Parkash V, Lindholm P, Peranen J, Kalkkinen N, Oksanen E, Saarma M, et al. The structure of the conserved neurotrophic factors MANF and CDNF explains why they are bifunctional. Protein Eng Des Sel 2009, 22: 233–241.
Hellman M, Arumae U, Yu LY, Lindholm P, Peranen J, Saarma M, et al. Mesencephalic astrocyte-derived neurotrophic factor (MANF) has a unique mechanism to rescue apoptotic neurons. J Biol Chem 2011, 286: 2675–2680.
Vimal P, PäIvi L, Johan PN, Nisse K, Esko O, Mart S, et al. The structure of the conserved neurotrophic factors MANF and CDNF explains why they are bifunctional. Protein Eng Des Sel 2009, 22: 233–241.
Lindahl M, Saarma M, Lindholm P. Unconventional neurotrophic factors CDNF and MANF: Structure, physiological functions and therapeutic potential. Neurobiol Dis 2017, 97: 90–102.
Lohoff FW, Bloch PJ, Ferraro TN, Berrettini WH, Pettinati HM, Dackis CA, et al. Association analysis between polymorphisms in the conserved dopamine neurotrophic factor (CDNF) gene and cocaine dependence. Neurosci Lett 2009, 453: 199–203.
Apostolou A, Shen Y, Liang Y, Luo J, Fang S. Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp Cell Res 2008, 314: 2454–2467.
Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem 2003, 278: 19367–19377.
Gang C, Bower KA, Cuiling M, Shengyun F, Thiele CJ, Jia L. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J 2004, 18: 1162–1164.
Yusuke S, Kohsuke T, Hidenori I. The ASK1-MAP kinase signaling in ER stress and neurodegenerative diseases. Curr Mol Med 2006, 6: 87–97.
Lindholm P, Saarma M. Novel CDNF/MANF family of neurotrophic factors. Dev Neurobiol 2010, 70: 360–371.
Lindstrom R, Lindholm P, Kallijarvi J, Yu LY, Piepponen TP, Arumae U, et al. Characterization of the structural and functional determinants of MANF/CDNF in Drosophila in vivo model. PLoS One 2013, 8: e73928.
Liu H, Zhao C, Zhong L, Liu J, Zhang S, Cheng B, et al. Key subdomains in the C-terminal of cerebral dopamine neurotrophic factor regulate the protein secretion. Biochem Biophys Res Commun 2015, 465: 427–432.
Choi JM, Hong JH, Chae MJ, Ngyuen PH, Kang HS, Ma HI, et al. Analysis of mutations and the association between polymorphisms in the cerebral dopamine neurotrophic factor (CDNF) gene and Parkinson disease. Neurosci Lett 2011, 493: 97–101.
Sun ZP, Gong L, Huang SH, Geng Z, Cheng L, Chen ZY. Intracellular trafficking and secretion of cerebral dopamine neurotrophic factor in neurosecretory cells. J Neurochem 2011, 117: 121–132.
Palgi M, Lindstrom R, Peranen J, Piepponen TP, Saarma M, Heino TI. Evidence that DmMANF is an invertebrate neurotrophic factor supporting dopaminergic neurons. Proc Natl Acad Sci U S A 2009, 106: 2429–2434.
Niles LP, Sathiyapalan A, Bahna S, Kang NH, Pan Y. Valproic acid up-regulates melatonin MT1 and MT2 receptors and neurotrophic factors CDNF and MANF in the rat brain. Int J Neuropsychopharmacol 2012, 15: 1343–1350.
Almutawaa W, Kang NH, Pan Y, Niles LP. Induction of neurotrophic and differentiation factors in neural stem cells by valproic acid. Basic Clin Pharmacol Toxicol 2014, 115: 216–221.
Lindholm P, Peranen J, Andressoo JO, Kalkkinen N, Kokaia Z, Lindvall O, et al. MANF is widely expressed in mammalian tissues and differently regulated after ischemic and epileptic insults in rodent brain. Mol Cell Neurosci 2008, 39: 356–371.
Wang H, Ke Z, Alimov A, Xu M, Frank JA, Fang S, et al. Spatiotemporal expression of MANF in the developing rat brain. PLoS One 2014, 9: e90433.
Tsybko AS, Ilchibaeva TV, Kulikov AV, Kulikova EA, Krasnov IB, Sychev VN, et al. Effect of microgravity on glial cell line-derived neurotrophic factor and cerebral dopamine neurotrophic factor gene expression in the mouse brain. J Neurosci Res 2015, 93: 1399–1404.
Tadimalla A, Belmont PJ, Thuerauf DJ, Glassy MS, Martindale JJ, Gude N, et al. Mesencephalic astrocyte-derived neurotrophic factor is an ischemia-inducible secreted endoplasmic reticulum stress response protein in the heart. Circ Res 2008, 103: 1249–1258.
Norisada J, Hirata Y, Amaya F, Kiuchi K, Oh-hashi K. A comparative analysis of the molecular features of MANF and CDNF. PLoS One 2016, 11: e0146923.
Glembotski CC, Thuerauf DJ, Huang C, Vekich JA, Gottlieb RA, Doroudgar S. Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J Biol Chem 2012, 287: 25893–25904.
Lu B. Pro-region of neurotrophins: role in synaptic modulation. Neuron 2003, 39: 735–738.
Huang J, Chen C, Gu H, Li C, Fu X, Jiang M, et al. Mesencephalic astrocyte-derived neurotrophic factor reduces cell apoptosis via upregulating GRP78 in SH-SY5Y cells. Cell Biol Int 2016, 40: 803–811.
Chen L, Feng L, Wang X, Du J, Chen Y, Yang W, et al. Mesencephalic astrocyte-derived neurotrophic factor is involved in inflammation by negatively regulating the NF-kappaB pathway. Sci Rep 2015, 5: 8133.
Zhou C, Xiao C, Commissiong JW, Krnjevic K, Ye JH. Mesencephalic astrocyte-derived neurotrophic factor enhances nigral gamma-aminobutyric acid release. Neuroreport 2006, 17: 293–297.
Voutilainen MH, Back S, Peranen J, Lindholm P, Raasmaja A, Mannisto PT, et al. Chronic infusion of CDNF prevents 6-OHDA-induced deficits in a rat model of Parkinson’s disease. Exp Neurol 2011, 228: 99–108.
Kemppainen S, Lindholm P, Galli E, Lahtinen HM, Koivisto H, Hamalainen E, et al. Cerebral dopamine neurotrophic factor improves long-term memory in APP/PS1 transgenic mice modeling Alzheimer’s disease as well as in wild-type mice. Behav Brain Res 2015, 291: 1–11.
Zhou W, Chang L, Fang Y, Du Z, Li Y, Song Y, et al. Cerebral dopamine neurotrophic factor alleviates Abeta25-35-induced endoplasmic reticulum stress and early synaptotoxicity in rat hippocampal cells. Neurosci Lett 2016, 633: 40–46.
Ma CH, Omura T, Cobos EJ, Latremoliere A, Ghasemlou N, Brenner GJ, et al. Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J Clin Invest 2011, 121: 4332–4347.
Shakhbazau A, Martinez JA, Xu QG, Kawasoe J, van Minnen J, Midha R. Evidence for a systemic regulation of neurotrophin synthesis in response to peripheral nerve injury. J Neurochem 2012, 122: 501–511.
Fernández A, Guzmán S, Cruz Y, Zamorano P. Construction of bicistronic lentiviral vectors for tracking the expression of CDNF in transduced cells. Plasmid 2014, 76c: 15–23.
Yi L, Lin N, Hua Z, Wen Z, Yuan-Qiang Z, Shuai-Shuai W, et al. Conserved dopamine neurotrophic factor-transduced mesenchymal stem cells promote axon regeneration and functional recovery of injured sciatic nerve. Plos One 2014, 9: e110993–e110993.
Cheng L, Liu Y, Zhao H, Zhang W, Guo YJ, Nie L. Lentiviral-mediated transfer of CDNF promotes nerve regeneration and functional recovery after sciatic nerve injury in adult rats. Biochem Biophys Res Commun 2013, 440: 330–335.
Garea-Rodriguez E, Eesmaa A, Lindholm P, Schlumbohm C, Konig J, Meller B, et al. Comparative analysis of the effects of neurotrophic factors CDNF and GDNF in a nonhuman primate model of Parkinson’s disease. PLoS One 2016, 11: e0149776.
Walker Z, Costa DC, Walker RW, Lee L, Livingston G, Jaros E, et al. Striatal dopamine transporter in dementia with Lewy bodies and Parkinson disease: a comparison. Neurology 2004, 62: 1568–1572.
Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 2014, 75: 351–362.
Hoffer BJ. Commentary on chronic infusion of CDNF prevents 6-OHDA-induced deficits in a rat model of Parkinson’s disease. Merja H. Voutilainen et al. Exp Neurol 2011, 230: 162–166.
Domanskyi A, Saarma M, Airavaara M. Prospects of neurotrophic factors for Parkinson’s disease: comparison of protein and gene therapy. Hum Gene Ther 2015, 26: 550–559.
Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J Leukoc Biol 2007, 82: 1083–1094.
Bohn MC. Motoneurons crave glial cell line-derived neurotrophic factor. Exp Neurol 2004, 190: 263–275.
Tomac A, Widenfalk J, Lin LF, Kohno T, Ebendal T, Hoffer BJ, et al. Retrograde axonal transport of glial cell line-derived neurotrophic factor in the adult nigrostriatal system suggests a trophic role in the adult. Proc Natl Acad Sci U S A 1995, 92: 8274–8278.
Rocha SM, Cristovao AC, Campos FL, Fonseca CP, Baltazar G. Astrocyte-derived GDNF is a potent inhibitor of microglial activation. Neurobiol Dis 2012, 47: 407–415.
Cheng L, Zhao H, Zhang W, Liu B, Liu Y, Guo Y, et al. Overexpression of conserved dopamine neurotrophic factor (CDNF) in astrocytes alleviates endoplasmic reticulum stress-induced cell damage and inflammatory cytokine secretion. Biochem Biophys Res Commun 2013, 435: 34–39.
Zhao H, Cheng L, Liu Y, Zhang W, Maharjan S, Cui Z, et al. Mechanisms of anti-inflammatory property of conserved dopamine neurotrophic factor: inhibition of JNK signaling in lipopolysaccharide-induced microglia. J Mol Neurosci 2014, 52: 186–192.
Radi E, Formichi P, Battisti C, Federico A. Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis 2014, 42 Suppl 3: S125–152.
Hosoi T, Ozawa K. Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clin Sci (Lond) 2010, 118: 19–29.
Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, David R, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci 2002, 22: 10690–10698.
Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ 2006, 13: 385–392.
Liu H, Tang X, Gong L. Mesencephalic astrocyte-derived neurotrophic factor and cerebral dopamine neurotrophic factor: New endoplasmic reticulum stress response proteins. Eur J Pharmacol 2015, 750: 118–122.
Mei JM, Niu CS. Effects of CDNF on 6-OHDA-induced apoptosis in PC12 cells via modulation of Bcl-2/Bax and caspase-3 activation. Neurol Sci 2014, 35: 1275–1280.
Mei J, Niu C. Protective and reversal effects of conserved dopamine neurotrophic factor on PC12 cells following 6-hydroxydopamine administration. Mol Med Rep 2015, 12: 297–302.
Sawada M, Sun W, Hayes P, Leskov K, Boothman DA, Matsuyama S. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol 2003, 5: 320–329.
Xu H, Belkacemi L, Jog M, Parrent A, Hebb MO. Neurotrophic factor expression in expandable cell populations from brain samples in living patients with Parkinson’s disease. FASEB J 2013, 27: 4157–4168.
Mei J, Niu C. Effects of engineered conserved dopamine neurotrophic factor-expressing bone marrow stromal cells on dopaminergic neurons following 6-OHDA administrations. Mol Med Rep 2015, 11: 1207–1213.
Jiaming M, Niu C. Comparing neuroprotective effects of CDNF-expressing bone marrow derived mesenchymal stem cells via differing routes of administration utilizing an in vivo model of Parkinson’s disease. Neurol Sci 2015, 36: 281–287.
Zhao H, Cheng L, Du X, Hou Y, Liu Y, Cui Z, et al. Transplantation of cerebral dopamine neurotrophic factor transducted BMSCs in contusion spinal cord injury of rats: promotion of nerve regeneration by alleviating neuroinflammation. Mol Neurobiol 2016, 53: 187–199.
Bartus RT, Baumann TL, Brown L, Kruegel BR, Ostrove JM, Herzog CD. Advancing neurotrophic factors as treatments for age-related neurodegenerative diseases: developing and demonstrating “clinical proof-of-concept” for AAV-neurturin (CERE-120) in Parkinson’s disease. Neurobiol Aging 2013, 34: 35–61.
Lim ST, Airavaara M, Harvey BK. Viral vectors for neurotrophic factor delivery: a gene therapy approach for neurodegenerative diseases of the CNS. Pharmacol Res 2010, 61: 14–26.
Ren X, Zhang T, Gong X, Hu G, Ding W, Wang X. AAV2-mediated striatum delivery of human CDNF prevents the deterioration of midbrain dopamine neurons in a 6-hydroxydopamine induced parkinsonian rat model. Exp Neurol 2013, 248: 148–156.
Fernandez A, Guzman S, Cruz Y, Zamorano P. Construction of bicistronic lentiviral vectors for tracking the expression of CDNF in transduced cells. Plasmid 2014, 76: 15–23.
Cordero-Llana O, Houghton BC, Rinaldi F, Taylor H, Yanez-Munoz RJ, Uney JB, et al. Enhanced efficacy of the CDNF/MANF family by combined intranigral overexpression in the 6-OHDA rat model of Parkinson’s disease. Mol Ther 2015, 23: 244–254.
Acknowledgments
This review was supported by the National Natural Science Foundation of China (31471114, 31500837, and 31540075), the Taishan Scholarship and Program for New Century Excellent Talents in University, and the Natural Science Foundation of Shandong Province, China (BS2015SW022).
Author information
Authors and Affiliations
Corresponding author
Additional information
Tingting Tang and Yong Li contributed equally to this review.
Rights and permissions
About this article

Cite this article
Tang, T., Li, Y., Jiao, Q. et al. Cerebral Dopamine Neurotrophic Factor: A Potential Therapeutic Agent for Parkinson’s Disease. Neurosci. Bull. 33, 568–575 (2017). https://doi.org/10.1007/s12264-017-0123-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-017-0123-4