
Abstract
Rosai-Dorfman Disease (RDD) is a rare disease of unknown aetiology, initially described as a separate entity in 1969 by Rosai and Dorfman. We describe here a case of RDD presenting with acute, recurrent lymphadenitis and massive lymphadenopathy. The patient’s good response to the antibiotics was a false pointer towards infective pathology. The repeated fine needle aspiration cytology was inconclusive or suggested nonspecific reactive hyperplasia. The excision biopsy confirmed the diagnosis of RDD and was supported by immune-histochemistry for S-100 antigen. No other lymph node group was involved clinically or radiologically. The patient was treated with long-term antibiotic and low-dose predenisolone. The residual lymph node mass was excised after 3 months of first surgery. The patient is on regular follow-up for last 2 years and no recurrence has been reported.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rosai-Dorfman disease (RDD) is a rare histiocytic disorder initially described as a separate entity in 1969 by Rosai and Dorfman under the term sinus histiocytosis with massive lymphadenopathy (SHML) [1]. Histologically, lymph nodes show pericapsular fibrosis and dilated sinuses, heavily infiltrated with large histiocytes, lymphocytes and plasma cells. The presence of emperipolesis, or the engulfment of lymphocytes and erythrocytes by histiocytes that express S-100, is considered diagnostic of RDD although not uniquely [2].
We are reporting a rare case of RDD in a 37-year-old male, who presented with recurrent acute lymphadenitis and our treatment experience.
Case Report
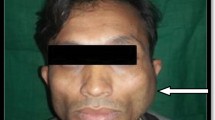
The 37-year-old male, Nepali citizen, staying in India for last 20 years, chef by occupation with no known comorbidities, presented with acute onset fever and painful swelling of the right submandibular region in Aug 2013. On local examination, there were multiple tender, enlarged and matted lymph nodes (LNs) of pre-auricular, level I and level II group on the right side, measuring approximately ∼03 × 04 cm. No other LN group was palpable in the neck and elsewhere. His routine workup, chest X-ray and the immune status were unremarkable except for leukocytosis and raised ESR. The ultrasonography neck confirmed the clinical findings with no involvement of the other LN groups. The clinical diagnosis of acute suppurative lymphadenitis with the differential diagnosis of tuberculosis was made. The patient was treated with antibiotics and analgesics. The patient responded well to treatment. Fine needle aspiration cytology (FNAC) was inconclusive or suggested nonspecific reactive hyperplasia on repetitions. Patient had three episodes of lymphadenitis in 5 months follow-up and one episode followed FNAC. The patient responded well to antibiotic treatment every time but the LNs remained massively enlarged. The excision biopsy was done under local anaesthesia of all involved LN groups except for one LN at level Ia due to dense pericapsular adhesions. Intra-op; there were multiple enlarged LNs with dense pericapsular adhesions. There was ∼0.5–1 ml thick pus inside the LNs hilum, pointing towards cold abscess.
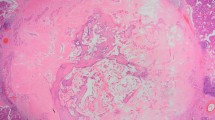
The HPE result was a surprise with the diagnosis of RDD (Fig. 1). As there are no clear cut guidelines for the treatment, the patient was treated with corticosteroids (predenisolone 60 mg once a day for 2 weeks). The predenisolone dose was tapered every 2 weeks by 10 mg. Low-dose predenisolone (10 mg, OD) was given for next 3 months. The USG scan for abdomen was normal with no evidence of intra-abdominal LAP. The patient remained asymptomatic except for level Ia LN enlargement. On the patient’s repeated requests and for fear of relapse, the same was excised too. The HPE confirmed again the diagnosis of RDD supported by IHC this time for S-100 antigen (Fig. 2). This time no corticosteroids were prescribed. The patient is on regular follow-up and no trouble has been reported so far.

Histopathology examination showing; a Partial effacement of lymph node architecture with dilated sinuses (haematoxylin & eosin stain, ×100). b Histiocytes showing engulfed lymphocytes and plasma cells—emperipolesis (haematoxylin & eosin stain, ×400)

Immunohistochemistry for S-100 antigen: a, b showing numerous histiocytes staining strongly for S-100 antigen protein with immune-reactivity score of 3+
Discussion
The most frequent clinical presentation of RDD is a massive bilateral and painless cervical lymphadenopathy with fever, night sweats and weight loss. Mediastinal, inguinal and retroperitoneal nodes may also be involved. Extra nodal involvement by RDD has been documented in 43 % of cases with the most frequent sites being the skin, soft tissue, upper respiratory tract, bone, eye and retro-orbital tissue with lymphadenopathy or as an isolated initial manifestation of disease [3].
Intracranial RDD usually occurs without extra cranial lymphadenopathy, and most intracranial lesions are attached to the dura with only few extending intra-parenchymally [4].
Patients with RDD without vital organ involvement should be followed closely without any active therapy. Patients with systemic symptoms or those with sudden enlargement of nodes may be treated with prolonged course of low-dose prednisone; the optimal duration of this is yet to be defined. For patients with vital organ compression, surgery and high-dose corticosteroids should be tried first, but radiotherapy may be needed in resistant cases or whenever surgery is not feasible [4].
Pulsoni et al. in their detail literature review had stressed that if RDD lesions are not massive and do not involve any vital organ, the patient should be observed only. In the presence of vital organ compression or other life-threatening manifestation surgical debulking and/or radiotherapy should be performed. Radiotherapy alone has proven ineffective in some patient. Chemotherapy has shown generally negative results [5].
This patient presented with recurrent lymphadenitis and massive LAP. The good response to antibiotics pointed towards infective pathology. In post-operative period, we have treated the patient with long-term antibiotics (1 month) and low-dose predenisolone (3 months) for which patient responded well. The antibiotics were added in view of pus present in LNs hilum. In spite of residual LAP, there was no lymphadenitis on follow up after first surgery. The patient’s initial presentation with acute lymphadenitis and involvement of few lymph node groups only was unique feature of this case. Further, the patient’s good response to antibiotics in preoperative period was a diagnostic dilemma further added by presence of pus in lymph nodes’ hilum.
References
Rosai J, Dorfman RF (1969) Sinus histiocytosis with massive lymphadenopathy. Arch Pathol 87:63–70
Paulli M, Bergamaschi G, Tonon L, Viglio A, Rosso R, Facchetti F, Geerts ML, Magrini U, Cazzola M (1995) Evidence for a polyclonal nature of the cell infiltrate in sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Br J Haematol 91:415–418
Foucar E, Rosai J, Dorfman R (1990) Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 7:19–73
Kraeft SK, Honig M, Krishnamurthy S (2007) Emperipolesis in the cerebrospinal fluid from a patient with Rosai-Dorfman disease. Diagn Cytopathol 36:67–68
Pulsoni A, Anghel G, Falcucci P, Matera R, Pescarmona E, Ribersani M, Villiva' N, Mandelli F (2002) Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): report of a case and literature review. Am J Hematol 69:67–71. doi:10.1002/ajh.10008
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Kumar, V., Bhandari, J.S., Awasthi, S. et al. Rosai-Dorfman Disease Presenting with Recurrent, Unilateral Submandibular Lymph Adenitis: a Case Report. Indian J Surg 78, 496–498 (2016). https://doi.org/10.1007/s12262-015-1440-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12262-015-1440-3