
Abstract
Purpose
Poor oral absorption, low bioavailability, short half-life, and gastrointestinal effects due to high dose of metformin required in the management of type-2 diabetes mellitus have spurred researchers to pay greater attention to the development of novel drug delivery systems to tackle these challenges. The aim of this study was to formulate and evaluate sunseed-oil-based PEGylated nanostructured lipid carriers (PEG-NLC) for enhanced delivery and prolonged antidiabetic activity of metformin.
Methods
The PEG-NLC and non-PEGylated NLC were formulated by high shear homogenization and thereafter characterized by scanning electron microscopy, mean particle size determination, photon correlation spectroscopy, differential scanning calorimetry (DSC), and Fourier transform infrared (FT-IR) spectroscopy. In vitro drug release, pharmacodynamic studies using alloxanized rat model, pharmacokinetics and safety evaluations were carried out. Results were compared with those of controls (market and pure samples of metformin).
Results
DSC results showed reduced crystallinity and hence greater possibility of enhanced drug solubility and entrapment, while FTIR results showed drug-excipient compatibility. The PEG-NLCs were safe, were stable spherical nanoparticles, had mean particle size, polydispersity indices and zeta potentials in the range of 290.6–880.6 nm, 0.494–0.625, and 26.1–32.8 mV, respectively. The PEG-NLCs showed enhanced drug release in simulated biorelevant media and prolonged antidiabetic activity compared with both non-PEGylated NLC and controls. Batch D40 containing the highest amount of PEG-4000 (optimized formulation) gave sixfold increase in pharmacokinetics properties than marketed sample (Glucophage®).
Conclusion
Sunseed-oil-based PEGylated NLC has proven to be a stable and safe carrier system for enhanced delivery and prolonged antidiabetic activity of metformin.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus is a chronic, complicated metabolic and endocrine disorder characterized by either absolute or relative lack of insulin due to destruction of the β cells of the islet of Langerhans, insufficient insulin production by the β cells of the islet of Langerhans, or resistance to the action of insulin [1]. The resultant effect of uncontrolled diabetes mellitus is hyperglycemia (fasting blood glucose > 126 mg/dl or > 7 mmol/L), leading to severe complications. It is the most typical endocrine–metabolic disorder characterized by chronic hyperglycemia giving rise to the risk of microvascular (retinopathy, nephropathy, and neuropathy) and macrovascular (ischemic heart disease, stroke, and peripheral vascular disease) damages, with associated reduced life expectancy and diminished quality of life [2]. According to the World Health Organization (WHO) statistics, the prevalence of diabetes has been rising more rapidly in middle- and low-income countries, and a majority of those living with diabetes live in low- and middle-income countries. The number of people living with diabetes has increased from 108 million in 1980 to 422 million in 2014, and the global prevalence of diabetes among adults over 18 years of age has risen from 4.7% in 1980 to 8.5% in 2014 [3]. In 2015, an estimated 1.6 million deaths were directly caused by diabetes, and another 2.2 million deaths were attributable to high blood glucose in 2012. Every six seconds, a person dies from diabetes, and 76% of diabetes-related deaths are in people under the age of 60 [4], and WHO projects that diabetes will be the seventh leading cause of death in 2030 [3]. A recent update by the International Diabetes Federation (IDF) atlas 2019, about 463 million adults (20–79 years) worldwide are living with diabetes, and this is projected to rise by about 51% to 700 million by 2045 [4]. According to the IDF, Africa, the region with the highest proportion of undiagnosed diabetes, albeit the lowest prevalence (3.9%), is projected to increase by 143.3% by 2045 [4].
Metformin hydrochloride is a biguanide and the most widely prescribed oral antihyperglycemic agent in managing type 2 diabetes mellitus [5]. However, despite its widespread clinical use, it is fraught with several problems such as poor absorption, low bioavailability, frequent dosage, high dose, short biological half-life, and gastrointestinal tract (GIT) side effects [6]. But by tactical engineering of lipid drug delivery systems, such as solidified reverse micellar solution (SRMS)-based solid lipid microparticles (SLMs) [5], nanostructured lipid carrier (NLCs), lipid drug conjugates (LDCs), and solid lipid nanoparticles (SLNs), these problems could be surmounted.
Attempts have been made to enhance the delivery of metformin; some of which includes the use of liposomes [7], PEGylated-mucin for oral delivery [8], alginate nanoparticles [9], hyaluronic acid nanoparticles [10], chitosan nanoparticles [11], chitosan/eudragit nanocomplex [12], niosomes [13], topical gel for derma delivery [14], solid lipid microparticles [5], solid lipid nanoparticle for transdermal delivery [15], reactive oxygen species (ROS)-responsive organosilica nanocarrier and metformin-pluronic-based polyurethanes for controlled drug delivery. The utilization of NLCs in drug delivery systems is because lipid matrices are safe, biocompatible, nontoxic, easily manufactured industrially, and versatile, thus bestowing its suitability for different routes of administration [16,17,18]. Nanostructured lipid carriers (NLCs) are second-generation lipid-based nanocarriers formed from a mixture of solid and liquid lipids and have unstructured matrix due to the different moieties of the constituents of NLCs [19]. The solid/liquid lipid blend ratio may vary from 70:30 to 99.9:0.1 [20,21,22]. The total lipid content of the NLC may vary from 5 to 40% [20,21,22,23], and the nanoparticles are stabilized in an aqueous medium using at least one surfactant in a concentration ranging from 0.5 to 5% w/w [5, 24, 25]. They were developed from solid lipid nanoparticles and have advantages over traditional lipid nanoparticles, such as improved drug incorporation and release properties [26]. In addition, they are more advantageous than any other nanoparticle preparation because of their oil-rich core, making for more significant drug entrapment [27]. Nanostructured lipid carriers came as a result of some of the failures/limitations of solid lipid nanoparticles, such as low payload, unpredictable gelation tendency, polymorphic transition, drug expulsion during storage [28], and high water content [17]. SLNs are prepared using solid lipids (i.e., lipids that are solid at room temperature as well as at body temperature) that are biocompatible and biodegradable [29]. The crystallinity of solid lipids affects the release properties of the SLNs derived [30], unlike in the matrix of NLCs, which is composed of a mixture of spatially different lipid molecules, which makes more imperfection in the matrix to accommodate more drug molecules than SLN [28, 31,32,33].
Davies and Abuchowsky first described PEGylation in the 1970s. It is the modification of a protein, peptide, or non-peptide molecule by linking one or more polyethylene glycol (PEG) chains. The polymer is usually nontoxic, non-immunogenic, non-antigenic, highly soluble in water, and FDA-approved [34]. It is a promising and extensively studied strategy for improving the performance of drugs and bioactives; it is mainly characterized by the use of very small quantities of substances for therapy [35]. The PEGylation of nanoparticles significantly changes a metabolic drug process in blood by protecting it from metabolism [36,37,38], conferring stealth properties on the nanoparticles [39], reducing the tendency of elimination by the reticuloendothelial system (RES), improving the bioavailability and half-life of drug used in the formulation [40]. PEG is hydrophilic; thus, its hydrophilic properties could prevent interaction with other nanoparticles via sterical hindrance, thereby improving nanoparticles’ stability [41]. PEGylated products have shown numerous advantages: a prolonged residence in the body, decreased degradation by metabolic enzymes and reduced or eliminated protein immunogenicity [34]. The novelty embodied in this study is the use of sunseed-oil-based PEGylated nanoengineered lipid carrier to enhance the antidiabetic activity of metformin via improved half-life, reduced dosage and dosing frequency as well as through enhanced permeability across the biological membrane. Although we had earlier utilized sunseed oil in the development of PEGylated formulations for enhanced delivery of antifungal agents[35], to the best of our knowledge, there is currently a dearth of information in the literature on the use of sunseed-oil-based PEGylated nanoengineered lipid carrier for oral delivery of metformin for enhanced treatment of diabetes mellitus. Therefore, the investigation of sunseed-oil-based PEGylated nanolipid carrier for enhanced metformin delivery to optimize the efficacy, intestinal absorption, bioavailability, biological half-life, and reduce drug dose, dosing frequency and drug adverse effects, informs the aim of this study. Thus, the objective of this study was to formulate, characterize and evaluate in vitroand in vivo pharmacodynamic properties of metformin-loaded sunseed-oil-based PEGylated nanostructured lipid carrier (PEG-NLC) for improved delivery of metformin hydrochloride.
Materials and Methods
Materials
Metformin hydrochloride pure sample was obtained as a gift from May and Baker PLC (Ikeja, Lagos State, Nigeria). Phospholipon® 90H (P90H) (Phospholipid GmbH, Köln, Germany), sorbitol (Caesar & Loretz, Hilden, Germany), polyethylene glycol 4000 (PEG 4000) (Ph. Eur. Carl Roth GmbH + Co. KG Karlsruhe, Germany), beeswax (Carl Roth, Karlsruhe, Germany), Polysorbate 80 (Tween® 80) (Acros Organics, Geel, Belgium), sunseed oil (double refined), vitamin A fortified (Kewalram Chanrai Group, Lagos, Nigeria), Alloxan (Merck KGaA, Darmstadt, Germany), distilled water (Lion water, University of Nigeria, Nsukka, Nigeria), and other solvents and reagents were used as procured from their manufacturers without further purification. Adult albino Wistar rats of both sexes were procured from the Faculty of Veterinary Medicine, University of Nigeria, Nsukka.
Preparation of PEGylated Lipid Matrices
PEGylated lipid matrices were prepared by the fusion method [42, 43] using beeswax (BW) and Phospholipon® 90H (P90H) (as solid lipids) in combination with super-refined sunseed oil (as liquid lipid) followed by PEGylation. The solid lipids and liquid lipid were used at a 7:3 ratio (i.e., 21.0 g of BW/P90H admixture and 9.0 g of super-refined sunseed oil). First, 21.0 g of beeswax and 9.0 g of P90H were weighed using an electronic balance (Mettler H8, Switzerland), poured into a crucible, and melted together at 70 °C on a thermoregulated oil bath (liquid paraffin) and stirred thoroughly to obtain an adequate mixing. Next, the homogenous mixture of the lipid matrix (LM1) was stirred further at room temperature and then allowed to cool and solidify. After 24 h, this lipid matrix was melted in the thermoregulated bath at a temperature of 80 °C, followed by the addition of 9.0 g (9.017 ml) of super-refined sunseed oil. The mixture was stirred continuously until a homogenous, transparent white melt was obtained. Next, the homogenous mixture of the lipid matrix (LM2) was stirred at room temperature until solidification. More so, after 24 h, various quantities (90, 80, and 60%w/w) of the prepared lipid matrix (LM2) were melted together with corresponding amounts of polyethylene glycol (PEG 4000) (10, 20, and 40%w/w) incorporated at 80 ºC over the oil bath to give PEGylated lipid matrices containing 1:9, 2:8, 4:6 ratios of PEG: lipid matrix, respectively, which were appropriately stirred and allowed to solidify.
Preparation of Drug-Loaded PEGylated Lipid Matrices
Representative drug-loaded PEGylated lipid matrices were prepared by fusion using the PEGylated lipid matrices and metformin. With target PEGylated lipid concentration of 5.0%w/w and target drug concentrations of 1.0%w/w of metformin in the PEGylated nanostructured lipid carriers to be developed, 2.5 g of each of the lipid matrices was melted in the thermoregulated oil bath at a temperature of 80 ºC followed by the addition of 0.5 g of metformin. Each mixture was stirred continuously until a homogenous, transparent white melt was obtained. Then, the drug-loaded lipid matrices were allowed to cool and solidify at room temperature.
Differential Scanning Calorimetry (DSC) Analysis of Plain and Drug-Loaded PEGylated Lipid Matrices
About 5 mg of each sample was weighed into an aluminum pan and hermetically sealed, and the thermal behavior was determined in the range of 20–350 °C at a heating rate of 5 °C/min. The temperature was held at 80 °C for 10 min and, after that, cooled at the rate of 5 to 10 °C/min. Baselines were determined using an empty pan, and all the thermograms were baseline-corrected.
Fourier Transform Infrared (FT-IR) Spectroscopic Analysis of Drug-Loaded PEGylated Lipid Matrices
FTIR spectroscopic analysis was conducted on metformin and representative metformin-loaded sunseed-oil-containing non-PEGylated and PEGylated beeswax-based P90H-modified lipid matrices (MT-loaded LM2 and MT-loaded PEG-LM2 or drug-loaded LM2 and drug-loaded PEG-LM2) using a Shimadzu FT-IR 8300 Spectrophotometer (Shimadzu, Tokyo, Japan). The spectrum was recorded in the wavelength region of 4000 to 400 cm−1 with a threshold of 1.303, sensitivity of 50, and resolution of 2 cm-1. A smart attenuated total reflection (SATR) accessory was used for data collection. The potassium bromate (KBr) plate used for the study was cleaned with a tri-solvent (acetone–toluene–methanol at 3:1:1 ratio) mixture for baseline scanning. A 0.1 g of each sample was mixed with 0.1 ml nujul diluent. The solution was introduced into the potassium bromate (KBr) plate and compressed into discs by applying a pressure of 5 tons for 5 min in a hydraulic press. The pellet was placed in the light path, and the spectrum was obtained. Spectra were collected in 60 s using Gram A1 spectroscopy software, and the chemometrics was performed using TQ Analyzer1.
Preparation of PEGylated Nanostructured Lipid Carriers
PEGylated nanostructured lipid carriers encapsulating metformin (D0, D10, D20, D40) were prepared using the drug, PEGylated lipid matrices, Polysorbate® 80 (Tween® 80) (mobile surfactant), sorbitol (cryoprotectant), and distilled water (vehicle) by the high shear hot homogenization method [44, 45]. In each case, the PEGylated lipid matrix was melted at 80 °C in the thermoregulated heater (IKA instrument), and metformin was introduced into the melted lipid and stirred thoroughly. Next, the aqueous phase containing sorbitol (4%w/w) and polysorbate® 80 at the same temperature was added to the molten lipid matrix with gentle stirring using a magnetic stirrer (SR 1 UM 52,188, Remi Equip., India), and the mixture was further dispersed using an Ultra-Turrax T25 (IKA-Werke, Staufen, Germany) homogenizer at 1000 rpm for 5 min. Finally, the obtained pre-emulsion was homogenized at 15,000 rpm for 30 min and allowed to cool and re-crystallize at room temperature. The formulation compositions of the PEGylated NLCs are shown in Tables 1 and 2.
Characterization of the Non-PEGylated and PEGylated NLCs
Determination of Particle Sizes, Polydispersity Indices and Surface Charges
Mean diameter, Z. Ave (nm), and polydispersity indices (PDI) of the formulations were measured using a zeta sizer nano-ZS (Malvern Instrument, Worcestershire, UK) equipped with a 10-mw He–NE laser employing the wavelength of 633 nm and a backscattering angle of 173° at 25 °C. To obtain a suitable scattering intensity, the sample was diluted with double-distilled water before photon correlation spectroscopic (PCS) analysis.
Furthermore, the stability of the formulations was ascertained using zeta potential measurement. Briefly, zeta potentials or surface charges (ζ) of metformin-loaded PEGylated and metformin-loaded non-PEGylated NLC formulations were estimated using dynamic light scattering (DLS) (Malvern Instruments, Japan). Each sample was diluted with deionized water to avoid multiple scattering and to maintain the number of counts per second in the region of 600, and measured at angle of 90° and temperature of 25 °C. Replicate measurements were taken and averages taken.
Compatibility Study by Fourier Transform Infrared (FTIR) Spectroscopy
Fourier transform infrared (FT-IR) spectroscopic analysis was conducted using a Shimadzu FT-IR 8300 Spectrophotometer (Shimadzu, Tokyo, Japan), and the spectrum was recorded in the wavelength region of 4000 to 400 cm−1 with a threshold of 1.303, the sensitivity of 50, and resolution of 2 cm−1 range. A smart attenuated total reflection (SATR) accessory was used for data collection. The potassium bromate (KBr) plate used for the study was cleaned with a tri-solvent (acetone–toluene–methanol at 3:1:1 ratio) mixture for baseline scanning. A 0.1 ml volume of each formulation was mixed with 0.1 ml nujul diluent. The solution was introduced into the potassium bromate (KBr) plate and compressed into discs by applying a pressure of 5 tons for 5 min in a hydraulic press. Then, the pellet was placed in the light path, and the spectrum was obtained. Spectra were collected in 60 s using Gram A1 spectroscopy software, and the chemometrics was performed using TQ Analyzer1.
Determination of Encapsulation Efficiency
The encapsulation efficiency of each formulation was determined. A 5 ml volume of each formulation was placed in a centrifuge tube and centrifuged for 30 min at an optimized speed of 4000 rpm to obtain two phases (the aqueous and lipid phases). A 1 ml volume of the aqueous phase was measured with the aid of a syringe and then diluted 10, 000-fold using distilled water. The absorbance of the dilutions was taken using a UV spectrophotometer (Jenway 6405, UK) at a wavelength of 231.5 nm, and the EE % was calculated using the formula below:
Loading Capacity
Loading capacity of the drug in lipid carriers depends on the type of lipid matrix, solution of drug in melted lipid, miscibility of drug melt and lipid melt, chemical and physical structure of solid lipid matrix, and the polymorphic state of the lipid material [42]. Therefore, loading capacity (LC) is expressed as the ratio between the entrapped drug by the lipid and the total quantity of the lipids used in the formulation. It was calculated using the formula below:
In Vitro Drug Release Studies
The USP XXII rotating paddle apparatus (Erweka, GmbH Germany) was employed for this release study. The dissolution medium consisted of 500 mL of freshly prepared phosphate buffer maintained at 37 ± 1 ℃ utilizing a thermoregulated water bath. The polycarbonate dialysis membrane used as a release barrier was pre-treated by soaking it in the dissolution medium for 24 h before the commencement of each release experiment. In each case, 2 ml of the drug-loaded PEGylated NLC was placed in the dialysis membrane, securely tied with a thermoresistant thread, and then immersed in the dissolution medium under agitation provided by the paddle at 200 rpm. At predetermined time intervals (30, 60, 120, 180, 240, 300, 360, 420, 480, and 600 min), 5 mL portions of the dissolution medium were withdrawn and replaced with an equal volume of the medium to maintain a sink condition, filtered with a pore size of 0.22 mm (Millipore filter, Delhi, India) and analyzed using a spectrophotometer (Jenway, UK) at 231.5 nm.
In Vivo Antidiabetic Studies
Experimental Animals
Wistar strain albino rats of both sexes weighing between 150 and 200 g were bred in our institution. The animals were housed in standard environmental conditions, kept at a body temperature of 37 °C using warming lamps, and left for one week to acclimatize with the new laboratory environment while being fed a standard laboratory low chow diet. All the animals were fasted for 12 h but were allowed free access to water before the commencement of the experiments.
Induction of Diabetes
Adult Wistar rats (either sex) were used to evaluate the antidiabetic effects of the formulations. The rats were divided into groups of six animals, and each group of animals was housed in a separate cage. Diabetes was induced by a single intraperitoneal injection of a freshly prepared solution of alloxan (150 mg/ kg) in normal saline for all the groups. After one h of alloxan administration, the animals were fed freely, and 5% dextrose solution was also given orally in a feeding bottle for a day to overcome the early hypoglycemic phase. The animals were kept under observation, and after 48 h, blood was withdrawn from the tail vein of the animals, and the blood glucose level was measured with a glucometer (Accu-check, Roche, USA). The diabetic rats (glucose level above 200 mg/dl) were separated into ten groups of six animals (n = 6).
Evaluation of Antidiabetic Activity
The different formulations of the metformin-loaded PEGylated NLC were administered orally to the animals according to their weight as follows:
-
Group A: received the test formulation (batch D0) equivalent to 100 mg/kg of metformin hydrochloride.
-
Group B: received the test formulation (batch D10) equivalent to 100 mg/kg of metformin hydrochloride.
-
Group C: received the test formulation (batch D20) equivalent to 100 mg/kg dose of metformin hydrochloride.
-
Group D: received the test formulation (batch D40) equivalent to 100 mg/kg dose of metformin hydrochloride.
-
Group E: received glucophage® equivalent to 100 mg/kg dose of metformin hydrochloride.
-
Group F: received pure metformin equivalent to 100 mg/kg dose of metformin hydrochloride.
-
Group G: received normal saline orally
All the samples for treatment were administered orally. In addition, blood samples of the animals were collected from the tail vein afterward at time intervals of 0, 1, 3, 6, 12, and 24 h and tested for blood glucose level using the glucometer.
The post-dose levels of the blood glucose were expressed as a percentage of the pre-dose level. The percent basal blood glucose concentration was plotted against time for the various groups.
In Vivo Bioavailability Study
In this study, eighteen (18) rats were used. They were made diabetics as described in the preceding section on antidiabetic study, after which the rats were randomly divided into three groups of six rats each. Group one received the MT-loaded non-PEGylated NLC formulation (batch G0) equivalent to 100 mg/kg dose of metformin hydrochloride, Group two received the MT-loaded PEGylated NLC formulation (batch G40) equivalent to 100 mg/kg dose of metformin hydrochloride, and Group three received Glucophage® (reference sample) equivalent to 100 mg/kg dose of metformin hydrochloride (positive control). The formulations were administered orally, and thereafter blood samples were collected from the tail vein of the rats at time intervals of 0, 1, 3, 6, 12, and 24 h using heparinized hematocrit tubes, centrifuged at 5,000 rpm for 5 min to separate the plasma and then stored at -4 ºC till analyzed. Approximately 0.2 ml of the plasma sample was diluted with equal volume of acetonitrile and centrifuged at 2,000 rpm for 5 min after which 0.1 ml of the supernatant was diluted in distilled water and assayed spectrophotometrically (Unico 2102 PC UV/Vis Spectrophotometer, New York, USA) for drug content. For the preparation of calibration curve, plasma from the blood withdrawn at zero hour was similarly diluted and used as blank. The plasma concentration–time curve was then plotted and further evaluated to obtain the pharmacokinetic parameters such as the maximum plasma concentration (Cmax) and the corresponding time (Tmax) using Phoenix® WinNonlin (version 6.3; Pharsight, St Louis, MO, USA), based on the average blood drug concentration. Area under the curve (AUC) from 0 to 24 h was calculated using the program’s linear trapezoidal rule.
Safety Studies
Here, three groups of animals were selected from the antidiabetic studies in the previous section, i.e., one animal from each group (optimized formulation group (G40), diabetic but untreated rat group and non-diabetic/normal rabbit group) was utilized for the purpose of histopathological studies in line with earlier reports [6, 8]. Small pieces of liver tissues in each group were collected in 10% neutral buffered formalin for proper fixation for 24 h. These tissues were processed and embedded in paraffin wax. Sections of 5–6 um in thickness were cut and stained with hematoxylin and eosin (H & E). These sections were examined photomicroscopically for necrosis, steatosis and fatty changes of hepatic cell [6, 8].
Statistical Analysis
All experiments were performed in replicate for the validity of the statistical analysis. Results were expressed as mean ± SEM. Analysis of variance (ANOVA) using LSD post hoc multiple comparison test was performed on the data sets generated using SPSS 16.0. Differences were considered significant for p-values < 0.05.
Results and Discussion
Thermal Properties of Plain and Drug-Loaded Lipid Matrices
Lipids used in nanoformulations have different crystallization temperatures, and these polymorphic transitions affect the nanoformulations’ thermal behaviors. The energies required to melt substances vary depending on whether crystalline or amorphous. Amorphous substances lack a long-range order or are less ordered; thus, lesser energy is needed to melt the substance; however, crystalline substances have a perfect crystalline structure and require higher energy to overcome lattice forces. Therefore, higher melting enthalpy values suggest a higher-ordered lattice arrangement [45]. The slight presence or total absence of a melting peak in the DSC of a solid dispersion indicates that the drug is partly or wholly amorphous/molecularly dispersed [46, 47]. The thermogram of beeswax, Phospholipon 90H, structured lipid matrix (LM1), structured lipid matrix (LM2), PEG 4000, PEGylated lipid matrix (LM2), metformin, metformin-loaded non-PEGylated (drug-loaded LM2), PEGylated lipid matrices (drug-loaded PEG-LM2), metformin-loaded non-PEGylated (drug-loaded LM2), and PEGylated lipid matrices (drug-loaded PEG-LM2) in superposition are shown in Figs. 1, 2, 3, 4, 5, 6, 7, 8, 9 and 10.

Differential scanning calorimetry (DSC) thermogram of beeswax (BW)

Differential scanning calorimetry (DSC) thermogram of Phospholipon® 90H (P90H)

Differential scanning calorimetry (DSC) thermogram of structured lipid matrix (BW: P90H) (7:3) (LM1)

Differential scanning calorimetry (DSC) thermogram of structured phospholipid-modified beeswax-based lipid matrix structured with sunseed oil (LM2)

Differential scanning calorimetry (DSC) thermogram of PEG 4000

Differential scanning calorimetry (DSC) thermogram of PEGylated lipid matrix (PEG-LM2)

Differential scanning calorimetry (DSC) thermogram of metformin

Differential scanning calorimetry (DSC) thermogram of metformin-loaded lipid matrix (drug-loaded LM2)

Differential scanning calorimetry (DSC) thermogram of metformin-loaded PEGylated lipid matrix (drug-loaded PEG-LM2)

DSC thermographs of metformin, metformin-loaded non-PEGylated and PEGylated lipid matrices (drug-loaded LM2) and (drug-loaded PEG-LM2) in superposition
The DSC thermograms of beeswax, PEG 4000, metformin, Phospholipon® 90H, and lipid matrix (LM1) showed single sharp endothermic melting peaks at 73, 74.5, 265.5, and 122.1 ℃ with enthalpies of -55.3, -3.8, -28.4, and -38.9 mW/mg, respectively, indicating the highly crystalline nature of these materials. Lipid matrix (LM1) and PEG lipid matrix (LM2) showed endothermic melting peaks at 88.6 and 93.1 ℃ with enthalpies of -32.5 and -3.9 mW/mg, respectively, and the reduced enthalpy indicates that the lipid matrices are less crystalline than the individual components used in the formulation. Lipid matrix (LM2), on the other hand, showed two peaks, an endothermic and an exothermic peak at 115.8 and 217.4 ℃ with enthalpy of -7.5 and 19.5 mW/mg; this indicates that lipid matrix (LM2) generates an imperfect matrix, which arises from a distortion of the crystal arrangement of the individual lipids after melting and solidification, thus creating more spaces for drug encapsulation [48, 49]. The exothermic peak at 217.4 ℃ could be due to the molecules arranging themselves to a lower energy configuration or chemical cure.
The DSC thermograms of metformin-loaded lipid matrix and metformin-loaded PEGylated lipid matrix (drug-loaded PEG-LM2) showed two melting peaks each. Although the metformin-loaded lipid matrix showed an endothermic peak at 94.5 and 258.0 ℃ with an enthalpy of -0.9 and -5.7 mW/mg, the melting peak was 258.0 ℃ is a weak peak, showing that metformin is either partly amorphous or molecularly dispersed in the lipid matrix. The metformin-loaded PEGylated lipid matrix (drug-loaded PEG-LM2) also showed two endothermic peaks at 89.0 and 258.0 ℃ with enthalpy of -0.7 and -0.4 mW/mg, indicating molecular dispersion of metformin in the lipid matrix. Furthermore, the DSC results illustrate that the drug-loaded lipid matrices had lower melting points than the pure metformin and did not show any transition of the drug in the thermogram, indicating that the drug exists in a solubilized form [50].
Fourier Transform Infrared Spectroscopy of Drug and Drug-Loaded Lipid Matrices
Solid-state characterizations are usually conducted to describe the properties of nanoformulations. The infrared (IR) spectrum of any given compound is always unique and characteristic of that compound [35]. FTIR spectroscopic analysis is usually carried out to evaluate possible molecular interactions between the drug and the matrices in the solid state [51]. The FTIR spectroscopic spectrum of metformin, metformin-loaded non-PEGylated (drug-loaded LM2), and PEGylated lipid matrices (drug-loaded PEG-LM2) are shown in Fig. 11. The pure metformin sample shows principal characteristic absorption bands at 3456.55 cm−1 (N–H stretching), 2931.90 cm−1 ((CH3)2-N absorption), 1643.41 cm−1 (N–H deformation), 1396.51 cm−1 (N–H deformation), 1219.05 cm−1 (C-O vibration), 1103.32 cm−1 (C-N stretching), 1130.74 cm−1 (C-N stretching), 771.56 cm−1 (N–H wagging), 601.81 cm−1 (C-H out of plane bending), and 547.8 cm−1 (C-N–C deformation). The spectrum of the metformin-loaded LM2 shows principal absorption bands at 3819.18 cm−1 (O–H stretching), 3471.98 cm−1 (N–H stretching), 2916.47 cm−1 ((CH3)2-N absorption), 2854.74 cm−1 ((CH3)2-N absorption), 2314.66 cm−1 (-C = C- stretching), 1735.99 cm−1 (-C = O vibration), 1643.41 cm−1 (N–H deformation), 1458.23 cm−1 (symmetric N–H deformation), 1396.51 cm−1 (N–H deformation), 1165.04 cm−1 (C-N stretching), 1095.60 cm−1 (C-N stretching), 1049.31 cm−1 (C-N stretching), 717.54 cm−1 (N–H wagging), 601.81 cm−1 (C-H out-of-plane bending) and 547.80 cm−1(C-N–C deformation). The spectrum of the PEGylated metformin-loaded lipid matrix (drug-loaded PEG-LM2) shows principal absorption bands at 3433.41 cm−1 (N–H stretching), 2916.47 cm−1 ((CH3)2-N absorption), 2854.74 cm−1 ((CH3)2-N absorption), 2314.66 cm−1 (-C = C- stretching), 1975.17 cm−1 (-C = O vibration), 1735.99 cm−1 (conjugated C = C bond vibration), 1651.12 cm−1 (N–H deformation), 1458.23 cm−1 (symmetric N–H deformation), 1226.77 cm−1 (C-O vibration), 1157.33 cm−1 (C-N stretching), 964.44 cm−1 (N–H out-of-plane bending), 840.99 cm−1 (NH2 rocking), 717.54 cm−1 (C-H out-of-plane bending), and 586.38 cm−1 (C-N–C deformation). From the results, it can be inferred that there was no interference between the drug, lipids, and polymers used in the formulation; this was seen from the fact that there was no significant change in the absorption peaks of metformin functional groups. The other absorption peaks present in the spectra were attributed to excipients; hence, the polymers and lipids used in the formulation are compatible with the drug.

FT-IR spectra of metformin, metformin-loaded non-PEGylated and PEGylated lipid matrices (drug-loaded LM2) and (drug-loaded PEG-LM2) in superposition
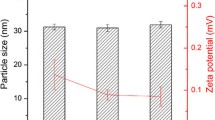
Mean Particle Size, Polydispersity Indices, and Surface Charges of the NLC Formulations
The combined particle size analysis and polydispersity index of the formulations are shown in Fig. 12. The formulation with the most miniature particle size was D20 (290.6 nm), and formulation D10 showed maximum particle size (880.60 nm). The PDI value of batch D0 and D10 was 0.477 and 0.625, showing the minimum and maximum PDI in the formulation, respectively. The particle size distribution was normal, and the polydispersity index confirmed this. These results showed a unimodal size distribution of the different particle sizes, indicating stable formulations [5]. Polydisperse systems tend to aggregate than monodisperse systems; thus, D40 is the most stable formulation (Table 3).

Combined particle size analysis result of the metformin-loaded sunseed-oil-based non-PEGylated and PEGylated nanostructured lipid carriers
The results of the zeta potential measurement surface charges as shown in Table 4 indicate good stability of the formulations since the zeta potential values were > 25 mV. The positive surface charges on the MT-loaded PEGylated and MT-loaded non-PEGylated NLC formulations imply that they would aid in easily transporting the formulations across cell membranes. In addition, results indicate that they can bind with negative charges on the mucosa walls of the GIT and ultimately prolong drug release for enhanced absorption, which is in agreement with the previous study [5].
Fourier Transform Infrared Spectroscopy of Drug-Loaded NLC Formulations
Figure 13 shows FTIR spectra of metformin, metformin-loaded non-PEGylated and PEGylated sunseed-oil-based nanostructured lipid carriers (D0, D10, D20, and D40) in superposition. The FTIR spectrum of the pure metformin sample shows characteristic principal absorption bands at 3456.55 cm−1 (N–H stretching), 2931.90 cm−1 ((CH3)2-N absorption), 1643.41 cm−1 (N–H deformation), 1396.51 cm−1 (N–H deformation), 1219.05 cm−1 (C-O vibration), 1103.32 cm−1 (C-N stretching), 1130.74 cm−1 (C-N stretching), 1130.74 cm−1 (C-N stretching), 771.56 cm−1 (N–H wagging), 601.81 cm−1 (C-H out-of-plane bending), and 547.8 cm−1 (C-N–C deformation). The FTIR spectrum of D0 shows principal absorption peaks at 3363.97 cm−1 (N–H stretching), 2090.91 cm−1 (carboxylic acid C = O vibration), 1643.41 cm−1 (N–H deformation), 1103.32 cm−1 (C-N stretching), and 578.66 cm−1 (C-N–C deformation). The FTIR spectrum of D10 shows principal absorption peaks at 3363.97 cm−1 (N–H stretching), 2106.34 cm−1 (carboxylic acid C = O vibration), 1635.69 cm−1 (N–H deformation), 1226.77 cm−1 (C-N stretching), 1080.17 cm−1 (C-N stretching), and 547.80 cm−1 (C-N–C deformation). The FTIR spectrum of D20 shows principal absorption peaks at 3363.97 cm−1 ((CH3)2-N absorption), 2924.18 cm−1, 2854.74 cm−1 (N–H stretching), 2121.77 cm−1 (carboxylic acid C = O vibration), 1643.41 cm−1 (N–H deformation), 1512.24 cm−1 (asymmetric N–H deformation), 1087.89 cm−1 (C-N stretching), and 532.37 cm−1 (C-N–C deformation). The FTIR spectrum of D20 shows principal absorption peaks at 3873.19 cm−1 (N–H stretching), 3302.24 cm−1 (asymmetric N–H stretching), 2924.18 cm−1, 2507.54 cm−1 ((CH3)2-N absorption), 2360.95 cm−1 (-C = C- stretching), 2083.19 cm−1 (carboxylic acid C = O vibration), 1620.26 cm−1 (N–H deformation), 1535.39 cm−1 (asymmetric N–H deformation), 1411.94 cm−1 (N–H deformation), 1033.88 cm−1 (C-N stretching), 794.7 cm−1 (N–H wagging) and 493.79 cm−1 (C-N–C deformation). From the results, it can be inferred that there was no interference in the drug, lipids, and polymers used in the formulation [52]; this was seen from the fact that there was no significant change in the absorption peaks of metformin functional groups, and the reduced peak intensities were due to the presence of hydrogen bonding [53]. The other absorption peaks present in the spectra were attributed to excipients; hence the polymers and lipids used in the formulation could be compatible with the drug.

FT-IR spectra of Metformin, Metformin-loaded non-PEGylated, and PEGylated sunseed-oil-based nanostructured lipid carriers (D0, D10, D20, and D40) in superposition
Scanning Electron Microscopy (SEM) of Drug-Loaded Non-PEGylated and PEGylated Nanostructured Lipid Carriers
Figure 14 shows the scanning electron micrograph of the NLC formulation for D40 (optimized formulation). The image showed the presence of well-defined nanodispersions with particles that were segregated, uniform in size, and semispherical in shape, with similar sizes spread over the formulation, which is the typical morphological aspect of nanoparticles.

Scanning electron micrograph (SEM) of metformin-loaded sunseed-oil-based PEGylated nanostructured lipid carrier (batch D40)
Encapsulation Efficiency and Loading Capacity of PEGylated and Non-PEGylated NLCs
The drug loading efficiency could be affected by the drug’s molecular weight, the volumetric size of the carrier, chemical interactions between the drug and the carrier, solubility of the drug in the carrier, miscibility in the lipid matrix, and the lipid phase polymorphic state [14, 17, 54]. Metformin has a high tendency to escape from the lipid matrix due to its high solubility in water [55] and may result in lower drug loading. When added in high concentration, PEG lipids may induce the formation of mixed micelles (PEGylated carriers), which may increase drug encapsulation efficiency [35]. This may account for the high encapsulation efficiency of the NLCs. Previous studies have shown a reverse relationship between particle size and loading parameters (loading efficiency and entrapment efficiency). Formulations with smaller particle sizes had higher encapsulation efficiency and drug loading capacity, and this is consistent with previous reports.
The encapsulation efficiency (EE%) and loading capacity (LC) of the NLCs are shown in Fig. 15. The encapsulation efficiencies were in the range of 98.78–99.83% for the NLCs, with the formulation D0 showing the highest EE%, while D20 had the least. However, all batches of the NLCs had good EE% (98.78–99.83%). The loading capacity of all the batches of NLCs showed a similar pattern. The loading capacity (LC) of the NLC batches was in the range of 16.50–16.64 g of metformin per 100 g of lipid. Again, the NLC batch D0 had the highest loading capacity, while D20 had the least.

Bar chart representation of encapsulation efficiency and drug loading capacity
In Vitro Drug Release From the Metformin-Loaded PEGylated and Non-PEGylated NLCs
The in vitro drug release study is a critical test to assess nanoparticle-based drug delivery systems’ safety, efficacy, and quality [56]. The in vitro release kinetics of nanoparticles provides vital information regarding the ability to modify the drug release; when performed correctly, they can be correlated to in vivo behaviors through predictive mathematical models [57, 58]. Many mathematical models have been proposed to predict release-kinetic models, such as the zero-order, first-order, Higuchi, Korsmeyer–Peppas, Weibull, Gompertz, Peppas–Sahlin, Bakers–Lonsdale, Hopfenberg, Hixson–Crowell [59]. Although there are numerous mathematical models, these models could not cover all possible release mechanisms [60, 61]. To identify a particular release mechanism, experimental data of statistical significance are compared to a solution of the theoretical model.
Predicting the release of the active ingredient from a formulation is a vital aspect of drug delivery. The release pattern is usually projected as a function of time using both sophisticated and straightforward mathematical models [62]. Kinetics is an essential factor in drug release studies because it provides information on drug concentration in plasma. Applying the kinetic model is critical in clarifying the release mechanism, which helps design the drug release control [4]. Five different mathematical equations were used to predict the release kinetics: zero-order equation, first-order equation, Hixson–Crowell, Higuchi, and Korsmeyer–Peppas models to describe the kinetics of metformin release from the PEG-NLC formulations. The most appropriate drug release model was selected based on the goodness-of-fit test. Although the formulations showed a strong correlation for the first order, Higuchi, Hixson–Crowell, zero-order, and Korsmeyer–Peppas had a good correlation coefficient (r2 ≥ 0.6899). The results showed that all the prepared formulations fit well with a strong correlation (r2 ≥ 0.9954), following the first-order kinetics. Therefore, the formulations predominantly followed the diffusion mechanism of drug release. The equation of first-order kinetics for batch D40 (representative sample) is:
Table 5 shows the values of the calculated squares of the correlation coefficient of the formulations for each model. The Korsmeyer–Peppas model showed the weakest correlation. According to this model, the determined release rate constants were between 0.7147 and 0.8336, and the coefficients ‘n’ of the equation did not exceed 1.0392 with the corresponding r2 values 0.6899–0.7337.
Figure 16 shows the release profiles of metformin from all the batches of PEG-NLC formulation in phosphate-buffered solution (PBS, pH 7.4). The in vitro release profiles of metformin in PBS indicate significantly (p < 0.05) greater release of metformin from optimized formulation (batch D40) when compared to the drug release from the marked formulation (Glucophage®). The different sets of the formulations showed a high release rate in the trend D40 = D0 > D10 > D20 > Glucophage®. The release rate of the drug was higher in the formulations than the release in Glucophage. Metformin was released in a sustained manner throughout the period of study. A similar drug release pattern was reported earlier [9]. This is expected as solid lipid nanoparticles slow down the mobility of drugs out of the particles [63]. A study by Xu et al. also showed an initial rapid release of metformin from lipid-based nanoformulations; this was predicted to be due to adsorption of metformin on the surface of the nanoparticles [64]. Also, as the concentration of PEG 4000 polymer used increases, the rate of drug release increases. Thus, PEGylation enhanced the dissolution and release rate of the drug from the formulations.

In vitro drug dissolution profile of metformin, metformin-loaded non-PEGylated and PEGylated sunseed-oil-based nanostructured lipid carriers (D0, D10, D20, and D40)
In Vivo Antidiabetic Activity
The results of the antidiabetic studies of the formulations, in comparison with the pure drug and commercially available product (glucophage®), are shown in Fig. 17 and Table 6. The effect of the PEGylated nanostructured lipid carrier (PEG-NLC) on the blood glucose level of non-diabetic rats was assessed and compared to both the pure drug sample and commercially available product (glucophage®). The in vivo pharmacologic antidiabetic activity of the formulations (D0, D10, D20, and D40), pure drug, and the commercially available product was evaluated using percentage reduction in blood glucose level (Table 6). This was the same evaluation method used by Mishra et al. where a percentage reduction in blood pressure was determined for Carvedilol-embedded nanostructured lipid carrier formulations [65]. The curves obtained by plotting percentage blood glucose reductions versus time are presented below. The groups treated with batch D0 and D10 formulations showed a higher and more sustained decrease in glucose level than those treated with the batch D20; there was a significant difference (p < 0.05) in the percentage reduction in blood glucose level between the groups that received formulations D0 and D10 with those that received D20. This could be due to the explanation that Shi et al. postulated in their study on the effects of polyethylene glycol on the surface of nanoparticles for targeted drug delivery. They said that encapsulation efficiency is an essential factor in the overall quality of the nanoparticles and that the PEG in nanoparticles often affect the encapsulation efficiency of the loaded molecules [66]. As the concentration of the PEG in the lipid fraction increases, the membrane's permeability also increases; this leads to leakage of the encapsulated drug, which negatively impacts the delivery of the drug [67,68,69]. The encapsulation efficiency of drugs that are soluble in water decreases as the PEG content of the lipid matrix increases. This negative relationship is also true for the encapsulation efficiency of water-soluble drugs and the molecular weight of the PEG in the lipid matrix, the probable reason being that the PEG will saturate the volume of the nanoparticles and the bigger the molecular weight of the PEG, the more the volume of the nanoparticles that are taken [70, 71]. Also, these batches produced a similar decrease in blood glucose level than the animal group treated with the market brand (glucophage®). This corroborated the results of Gadad et al. which showed that the rosuvastatin embedded nanostructured lipid carriers improved its bioavailability by two-fold compared to the marketed product [72].

Graphical representation of the in vivo antidiabetic effect (blood glucose-lowering effect) of the formulations (D0, D10, D20, and D40) vis-à-vis pure metformin, glucophage®, and distilled water
Furthermore, in the animal groups treated with the batches D0, D10, D20, and D40, the blood glucose reduction effectively commenced within an hour of oral administration with maximum blood glucose reduction (Tmax) achieved in 24 h. The blood glucose level produced by D0, D10, and D40 formulations reduced from 100% to 44.36%, 43.51%, and 44.41%, respectively, indicating that batch D40 was the best formulation, which was optimized for further in vivo bioavailability study. However, the blood glucose levels did not return to the initial level at zero hours within 24 h of the study.
In comparison with the pure drug and commercial sample, the formulations generally maintained the blood glucose level of the rats within the normoglycemic level for 12–24 h. Thus, it depicts that metformin could be effectively and efficiently delivered as sunseed-oil-based PEGylated NLCs. This also corresponded with the results of Gadad et al. which showed that the Rosuvastatin-embedded nanostructured lipid carrier was able to provide a sustained release of the drug over an extended period [68]. It can also be extrapolated that the blood-glucose-lowering effect was dependent on the polymers used and their concentrations; however, as the concentration of the PEG increases, this observed effect tends to decrease. The improved performance observed in the animal groups treated with the formulations corresponds with the improved dissolution observed.
In Vivo Oral Bioavailability
The pharmacokinetics results are shown in Fig. 18 and Table 7 as plasma concentration–time profiles of metformin and metformin-loaded non-PEGylated and PEGylated NLC and pharmacokinetic data, respectively. The mean AUC-24 values for optimized metformin-loaded non-PEGylated and PEGylated NLC indicate approximately threefold and sixfold increase in systemic bioavailability of metformin from metformin-loaded non-PEGylated and PEGylated NLC, respectively. Similarly, the mean Cmax values for non-PEGylated and PEGylated NLC formulations were significantly (p < 0.005) greater than that for the marketed sample.

Changes of metformin concentration in blood over 24-h study period, of rats orally administered with the optimized metformin-loaded PEGylated NLC (G40) and metformin-loaded non-PEGylated NLC (G0) in comparison with marketed formulation (Glucophage®) at equivalent dose
Obviously, the developed formulations recorded greater increased at broader peaks of mean plasma concentrations than the marketed metformin formulation (Glucophage®, reference sample) after an oral administration, implying that plasma concentration of metformin obtained with the reference sample was not sustained compared with the developed formulations. Thus, metformin-loaded non-PEGylated and PEGylated NLC formulations maintained a steady slow decrease or gradual clearance of drug throughout the study compared with the rapid exponential decrease in the marketed sample (Glucophage®). The decrease in metformin concentration in the blood by the formulation could be attributed to an increase in the circulating half-life of metformin when administered as metformin-loaded PEGylated NLC. Thus, the PEGylated nature of batch G40 further delayed the clearance of metformin from the blood circulation. By implication, there was enhancement in the circulation longevity of metformin in the developed NLC formulations, but the effect was highest with metformin-loaded PEGylated NLC. This implies that lower doses of the drug in nanoscale (PEGylated NLC) would be employed at reduced dosing frequency to achieve the desired antidiabetic effect of metformin with reduced unwanted side effects, which is consistent with earlier reports [6, 8].
Safety of the Metformin-Loaded PEGylated NLC Formulations
The effects of the optimized formulation on the liver of the rats are shown in Fig. 19. The observation from the histological studies conducted on the liver of the rabbits from various groups showed significant hepatitis (arrow) on the liver of the rats in diabetic but untreated group (b). Photomicrograph of liver section of rats from experimental diabetic group that received metformin-loaded PEGylated NLC (optimized batch D40) (a) showed reduced periportal mononuclear infiltration of cells-periportal hepatitis (arrow), while the non-diabetic and untreated group (c) showed normal portal area and hepatocytes (H and E, mag. × 400). Overall, the results of histological study showed that short-term administration of developed metformin-loaded PEGylated NLC formulation did not cause any significant changes in the body regarding the relative liver morphology of the treated rats. By implication, short-term oral administration of sunseed-oil-based metformin-loaded PEGylated NLC will have no negative effects on somatic growth.

Photomicrographs of liver section of rats from experimental diabetic group that received metformin-loaded PEGylated NLC (optimized batch D40) (a), liver section of the rats in diabetic but untreated group (b) and liver of the rats in the non-diabetic and untreated group (c)
Conclusions
This study evaluates the potential of metformin-loaded beeswax-based PEGylated nanostructured lipid carrier (PEG-NLC) for enhanced metformin delivery to treat diabetes mellitus. Metformin-loaded beeswax-based PEGylated nanostructured lipid carriers (PEG-NLCs) were successfully prepared by high shear hot homogenization method using structured lipids (beeswax and Phospholipon 90H), liquid lipids (sunseed oil), and PEG-4000 and evaluated for improved treatment of diabetes mellitus. Solid-state characterization performed on the lipid matrices used in preparing the formulations confirmed the suitability of the lipid matrices and their compatibility with metformin. There was sustained release of metformin from the PEGylated NLC formulation, which gave rise to enhanced antidiabetic activity and contributed to the circulation longevity of metformin in the blood, with formulation containing the highest amount of PEG-4000 (batch D40) (optimized formulation) recording sixfold increase in AUC than marketed sample (Glucophage®). The study suggests that nanostructured lipid carriers of metformin are suitable carriers for the oral delivery of metformin and other hydrophilic drugs. Using sunseed-oil-based PEGylated NLC as a delivery system for metformin achieves a steady-state drug concentration in the blood that is therapeutically effective, safe, and remains for an extended period. Sunseed-oil-based PEG-NLCs present a promising approach for improving metformin delivery and oral bioavailability.
References
Shi F, Wei Z, Zhao Y, Xu X. Nanostructured lipid carriers loaded with baicalin: An efficient carrier for enhanced antidiabetic effects. Pharmacogn Mag. 2016;12(47):198–202.
Heydari I, Radi V, Razmjou S, Amiri A. Chronic complications of diabetes mellitus in newly diagnosed patients. Int J Diabet Mellitus. 2010;2(1):61–3.
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442.
Suvi K, Belma M, Pouya S, Paraskevi S, editors. IDF Diabetes Atlas. 9th ed. [Internet]. International Diabestes Federation; 2019. Available from: http://www.diabetesatlas.org. Accessed 14 Jun 2020.
Momoh M, Kenechukwu F, Attama A. Formulation and evaluation of novel solid lipid microparticles as a sustained release system for the delivery of metformin hydrochloride. Drug Deliv. 2013;20(3–4):102–11.
Momoh M, Adikwu M, Ibezim E, Attama A. Effect of metformin and Vernonia amygdalina leaf extract loaded PEGylated-mucin formulation on haematological, kidney and liver indices of healthy and diabetes rats. J Pharm Res. 2011;4(10):3455–9.
Shukla SK, Kulkarni NS, Chan A, Parvathaneni V, Farrales P, Muth A, et al. Metformin-encapsulated liposome delivery system: An effective treatment approach against breast cancer. Pharmaceutics. 2019;11(11). http://www.ncbi.nlm.nih.gov/pmc/articles/PMC6920889/. Accessed 14 Jun 2020.
Momoh M, Adedokun M, Adikwu M, Kenechukwu F, Ibezim E, Ugwoke E. Design, characterization and evaluation of PEGylated-mucin for oral delivery of metformin hydrochloride. Afr J Pharm Pharmacol. 2013;7(7):347–55.
Kumar S, Bhanjana G, Verma RK, Dhingra D, Dilbaghi N, Kim K-H. Metformin-loaded alginate nanoparticles as an effective antidiabetic agent for controlled drug release. J Pharm Pharmacol. 2017;69(2):143–50.
Bhujbal S, Dash AK. Metformin-loaded hyaluronic acid nanostructure for oral delivery. AAPS Pharm Sci Tech. 2018;19(6):2543–53.
Santhosh C, Deivasigamani K, Venkata R. Enhanced effects of metformin loaded chitoson nanoparticles in L6 myotubes: In vitro evaluation. Pharm Lett. 2017;9(7):48–63.
Sahu AK, Verma A. Development and statistical optimization of chitosan and eudragit based gastroretentive controlled release multiparticulate system for bioavailability enhancement of metformin HCl. J Pharm Investig. 2016;46(3):239–52.
Hasan AA, Madkor H, Wageh S. Formulation and evaluation of metformin hydrochloride-loaded niosomes as controlled release drug delivery system. Drug Delivery. 2013;20(3–4):120–6.
Rostamkalaei SS, Akbari J, Saeedi M, Morteza-Semnani K, Nokhodchi A. Topical gel of metformin solid lipid nanoparticles: A hopeful promise as a dermal delivery system. Colloids Surf, B. 2019;175:150–7.
Sharma R, Sharma N, Rana S, Shivkumar H. Solid lipid nanoparticles as a carrier of metformin for transdermal delivery. Int J Drug Deliv. 2013;5:137–45.
Adhikari P, Pal P, Das AK, Ray S, Bhattacharjee A, Mazumder B. Nano lipid-drug conjugate: An integrated review. Int J Pharm. 2017;529(1–2):629–41.
Attama A, Momoh M, Builders P. Lipid nanoparticulate drug delivery systems: A revolution in dosage form design and development. In: Sezer AD, editor. Recent Advances in Novel Drug Carrier Systems [Internet]. Rijeka,Croatia: InTech; 2012. p. 107–40. Available from: http://www.intechopen.com/books/recent-advances-in-novel-drug-carrier-systems/lipid-nanoparticulate-drug-delivery-systems-a-revolution-in-dosage-form-design-and-development. Accessed 9 Jun 2021.
Chime SA, Onyishi IV. Lipid-based drug delivery systems (LDDS): Recent advances and applications of lipids in drug delivery. Afr J Pharm Pharmacol. 2013;7(48):3034–59.
Beloqui A, Solinís MÁ, Rodríguez-Gascón A, Almeida AJ, Préat V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine. 2016;12(1):143–61.
Doktorovová S, Kovačević Ab, Garcia Ml, Souto EB. Preclinical safety of solid lipid nanoparticles and nanostructured lipid carriers: Current evidence from in vitro and in vivo evaluation. Eur J Pharm Biopharm. 2016;108. Available from: https://pubmed.ncbi.nlm.nih.gov/27519829/ [cited 30 Jan 2022]
Carbone C, Teixeira MD, Sousa MD, Martins-Gomes C, Silva AM, Souto EM, et al. Clotrimazole-loaded Mediterranean essential oils NLC: A synergic treatment of candida skin infections. Pharmaceutics. 2019;11(5):231.
Teixeira MC, Carbone C, Souto EB. Beyond liposomes: Recent advances on lipid based nanostructures for poorly soluble/poorly permeable drug delivery. Prog Lipid Res. 2017;68:1–11.
Severino P, Andreani T, Macedo AS, Fangueiro JF, Santana MHA, Silva AM, et al. Current state-of-art and new trends on lipid nanoparticles (SLN and NLC) for oral drug delivery. J Drug Deliv. 2012;12: 750891.
Doktorovova S, Shegokar R, Souto E. Role of excipients in formulation development and biocompatibility of lipid nanoparticles (SLNs/NLCs). In: Grumezescu A, editor. Nanostructures for novel therapy. Amsterdam, The Netherlands: Elsevier; 2017. p. 811–43.
Pardeike J, Hommoss A, Müller Rh. Lipid nanoparticles (SLN, NLC) in cosmetic and pharmaceutical dermal products. Int J Pharm. 2009;366(1–2):170–84.
Zhang T, Chen J, Zhang Y, Shen Q, Pan W. Characterization and evaluation of nanostructured lipid carrier as a vehicle for oral delivery of etoposide. Eur J Pharm Sci. 2011;43(3):174–9.
Nnamani PO, Hansen S, Windbergs M, Lehr C-M. Development of artemether-loaded nanostructured lipid carrier (NLC) formulation for topical application. Int J Pharm. 2014;477(1–2):208–17.
Jaiswal P, Gidwani B, Vyas A. Nanostructured lipid carriers and their current application in targeted drug delivery. Artificial Cells, Nanomedicine, and Biotechnology. 2016;44(1):27–40.
Das S, Ng WK, Tan RBH. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): Development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur J Pharm Sci. 2012;47(1):139–51.
Iqbal MA, Md S, Sahni Jk, Baboota S, Dang S, Ali J. Nanostructured lipid carriers system: recent advances in drug delivery. J Drug Target. 2012;20(10). Available from: https://pubmed.ncbi.nlm.nih.gov/22931500/ [cited 30 Jan 2022]
Kaur K, Nautiyal U, Singh D. Nanostructured lipid carrier for bioavailability enhancement. Int J Recent Adv Sci Technol. 2015;2:1–7.
Fang C-L, Al-Suwayeh SA, Fang J-Y. Nanostructured lipid carriers (NLCs) for drug delivery and targeting. Recent Pat Nanotechnol. 2013;7(1):41–55.
Rajalakshmi G, Dhanapal CK, Sundhararajan R. An insight to nanostructured lipid carrier system. J Drug Deliv Therap. 2020;10:173–82.
Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today. 2005;10(21). Available from: https://pubmed.ncbi.nlm.nih.gov/16243265/ [cited 30 Jan 2022]
Kenechukwu FC, Attama AA, Ibezim EC, Nnamani PO, Umeyor CE, Uronnachi EM, et al. Novel intravaginal drug delivery system based on molecularly PEGylated lipid matrices for improved antifungal activity of miconazole nitrate. BioMed Res Int. 2018;20:1–18.
van der Meel R, Sulheim E, Shi Y, Kiessling F, Mulder WJM, Lammers T. Smart cancer nanomedicine. Nat Nanotechnol. 2019;14(11):1007–17.
Chen Q, Liu G, Liu S, Su H, Wang Y, Li J, et al. Remodeling the tumor microenvironment with emerging nanotherapeutics. Trends Pharmacol Sci. 2018;39(1):59–74.
Luo C, Sun J, Du Y, He Z. Emerging integrated nanohybrid drug delivery systems to facilitate the intravenous-to-oral switch in cancer chemotherapy. J Control Rel. 2014;176:94–103.
Nag OK, Awasthi V. Surface engineering of liposomes for stealth behavior. Pharmaceutics. 2013;5(4):542–69.
Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99:28–51.
Dai Y, Xing H, Song F, Yang Y, Qiu Z, Lu X, et al. Biotin-conjugated multilayer poly [D, L-lactide-co-glycolide]-lecithin-polyethylene glycol nanoparticles for targeted delivery of doxorubicin. J Pharm Sci. 2016;105(9):2949–58.
Barenholz Y, Anselem S. Quality control assays in the development and clinical use of liposome-based formulation. In Gregoriades G (ed): CRC Press, Boca Raton, FL; 1993. p. 527–616.
Eldridge J, Staas J, Meulbroek J, McGhee J, Tice T, Gilley R. Biodegradable microspheres as a vaccine delivery system. Mol immunol. 1991;28:287–94.
Pingale A, Gondkar S, Saudagar R. Nanostructured lipid carrier (NLC): A modern approach for intranasal drug delivery. World J Pharm Res. 2018;7(9):1574–88.
Uner M. Preparation, characterization and physico-chemical properties of solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC): Their benefits as colloidal drug carrier systems. Pharmazie. 2005;61:375–86.
Reginald-Opara JN, Attama A, Ofokansi K, Umeyor C, Kenechukwu F. Molecular interaction between glimepiride and Soluplus® -PEG 4000 hybrid based solid dispersions: Characterisation and anti-diabetic studies. Int J Pharm. 2015;496(2):741–50.
Kerc J, Srcic S. Thermal analysis of glassy pharmaceuticals. Thermochim Acta. 1995;248:81–95.
Attama AA, Nkemnele MO. In vitro evaluation of drug release from self micro-emulsifying drug delivery systems using a biodegradable homolipid from Capra hircus. Int J Pharm. 2005;304(1–2):4–10.
Uronnachi E, Ogbonna J, Kenechukwu F. Formulation and release characteristics of zidovudine-loaded solidified lipid microparticles. Trop J Pharm Res. 2014;13(2):199–204.
Uronnachi E, Ogbonna J, Kenechukwu F, Mumuni M, Attama A, Okore V. Pharmacokinetics and biodistribution of zidovudine loaded in a solidified reverse micellar delivery system. Int J Drug Deliv. 2013;5:73–80.
Tsutsumi S, Iida M, Tada N, Kojima T, Ikeda Y, Moriwaki T, et al. Characterization and evaluation of miconazole salts and cocrystals for improved physicochemical properties. Int J Pharm. 2011;421(2):230–6.
Kamboj VK, Verma PK. Preparation and characterization of metformin-loaded stearic acid coupled F127 nanoparticles. Asian J Pharm Clin Res. 2018;11(8):212–7.
Javidfar S, Pilehvar-Soltanahmadi Y, Farajzadeh R, Lotfi-Attari J, Shafiei-Irannejad V, Hashemi M, et al. The inhibitory effects of nano-encapsulated metformin on growth and hTERT expression in breast cancer cells. J Drug Deliv Sci Technol. 2018;43:19–26.
Sharma G, Parchur AK, Jagtap JM, Hansen CP, Joshi A. Hybrid nanostructures in targeted drug delivery. In: Hybrid Nanostructures for Cancer Theranostics. Elsevier; 2019. p. 139–58. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128139066000081. Accessed 26 Feb 2021.
Wadher KJ, Kakde RB, Umekar MJ. Development of a sustained-release tablet of metformin hydrochloride containing hydrophilic eudragit and ethyl cellulose polymer. Int J Clin Pharm. 2011;2(5):1–6.
Weng J, Tong HHY, Chow SF. In vitro release study of the polymeric drug nanoparticles: Development and validation of a novel method. Pharmaceutics. 2020;12(8):732.
Cardot J, Beyssac E, Alric M. In vitro–in vivo correlation: Importance of dissolution in IVIVC. Dissolution Technologies. 2007;14.
Sjögren E, Abrahamsson B, Augustijns P, Becker D, Bolger M, Brewster M, et al. In vivo methods for drug absorption - Comparative physiologies, model selection, correlations with in vitro methods (IVIVC), and applications for formulation/API/excipient characterization including food effects. Eur J Pharm Sci. 2014;57:23–31.
Bruschi ML, editor. Mathematical models of drug release. In: Strategies to Modify the Drug Release from Pharmaceutical Systems. Woodhead Publishing; 2015. p. 63–86. Available from: https://www.sciencedirect.com/science/article/pii/B9780081000922000059. Accessed 12 Aug 2019.
Khan MA, Shefeeq T. Role of mathematical modeling in controlled drug delivery. J Sci Res. 2009;1(3):539–50.
Permanadewi I, Kumoro A, Wardhani D, Aryanti N. Modelling of controlled drug release in gastrointestinal tract simulation. J Phys: Conf Ser. 2019;1295:1–8.
Siepmann J, Peppas N. Higuchi equation: Derivation, applications, use and misuse. Int J Pharm. 2011;418:6–12.
Ngwuluka NC, Kotak DJ, Devarajan PV. Design and characterization of metformin-loaded solid lipid nanoparticles for colon cancer. AAPS PharmSciTech. 2017;18(2):358–68.
Xu Q, Zhu T, Yi C, Shen Q. Characterization and evaluation of metformin-loaded solid lipid nanoparticles for celluar and mitochondrial uptake. Drug Dev Ind Pharm. 2016;42(5):701–6.
Mishra A, Imam SS, Aqil M, Ahad A, Sultana Y, Ameeduzzafar, et al. Carvedilol nano lipid carriers: formulation, characterization and in-vivo evaluation. Drug Deliv. 2016;23(4):1486–94.
Shi L, Zhang J, Zhao M, Tang S, Cheng X, Zhang W, et al. Effects of polyethylene glycol on the surface of nanoparticles for targeted drug delivery. Nanoscale. 2021;13(24):10748–64.
Hashizaki K, Taguchi H, Itoh C, Sakai H, Abe M, Saito Y, et al. Effects of poly(ethylene glycol) (PEG) concentration on the permeability of PEG-grafted liposomes. Chem Pharm Bull. 2005;53(1):27–31.
Edwards K, Almgren M. Surfactant-induced leakage and structural change of lecithin vesicles: effect of surfactant headgroup size. Langmuir. 1992;8(3):824–32.
Nicholas A., Scott M, Kennedy N, Jones M. Effect of grafted PEG-2000 on the size and permeability of vesicles. Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism. 1996;1304(2):120–8.
De Leo V, Ruscigno S, Trapani A, Di Gioia S, Milano F, Mandracchia D, et al. Preparation of drug-loaded small unilamellar liposomes and evaluation of their potential for the treatment of chronic respiratory diseases. Int J Pharm. 2018;545(1–2):378–88.
Lin T-T, Gao D-Y, Liu Y-C, Sung Y-C, Wan D, Liu J-Y, et al. Development and characterization of sorafenib-loaded PLGA nanoparticles for the systemic treatment of liver fibrosis. J Control Rel. 2016;221:62–70.
Gadad AP, Tigadi SG, Dandagi PM, Mastiholimath VS, Bolmal UB. Rosuvastatin-loaded nanostructured lipid carrierj for enhancement of oral bioavailability. Indian J Pharm Ed Res. 2016;50(4):605–11.
Funding
This research work received financial support from the Tertiary Education Trust Fund (TETFund) (Grant no. TETFUND/DR&D/CE/NRF/2019/STI/46/) by Government of Nigeria. Dr. Franklin C. Kenechukwu wishes to acknowledge Phospholipid GmbH, Köln, Germany, for generous provision of Phospholipon® 90H (P90H), Ph. Eur. Carl Roth GmbH + Co. KG Karlsruhe, Germany, for the king gift of polyethylene glycol 4000 (PEG 4000) and beeswax.
Author information
Authors and Affiliations
Contributions
Franklin Chimaobi Kenechukwu did conceptualization, supervision, methodology, validation, resources, funding acquisition, writing—original and draft as well as revision; Daniel Okwudili Nnamani performed methodology, writing—original and draft as well as revision; Bright Ugochukwu Nmesirionye was involved in methodology, formal analysis, investigation, writing—review and editing; God’spower Tochukwu Isaac contributed to methodology and writing—review and editing; Mumuni Audu Momoh and Anthony Amaechi Attamadone methodology, validation, resources, supervision, writing—review and editing.
Corresponding author
Ethics declarations
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All experimental protocols were conducted with strict adherence to the guidelines of the Institutional Animal Care and Use Committee of the University of Nigeria, Nsukka. Ethical clearance approval for in vivo antidiabetic studies was sought and obtained from the Faculty of Pharmaceutical Sciences Research Ethics Committee (UNN/FPS/2019-2020_017X) before the commencement of the in vivo animal studies.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kenechukwu, F.C., Nnamani, D.O., Nmesirionye, B.U. et al. PEGylated Lipid Nanocontainers Tailored with Sunseed-Oil-Based Solidified Reverse Micellar Solution for Enhanced Pharmacodynamics and Pharmacokinetics of Metformin. J Pharm Innov 18, 437–460 (2023). https://doi.org/10.1007/s12247-022-09654-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12247-022-09654-w