
Abstract
Purpose
Antibiotic-loaded polymethylmethacrylate has been conventionally used for osteomyelitis treatment, but it requires a second operation for removal. This study aimed to develop and examine the properties of a novel vancomycin-loaded composite fabricated from biodegradable poly(ε-caprolactone)/poly(hydroxybutyrate-co-hydroxyvalerate)/biphasic calcium phosphate (PCL/PHBV/BCP) for chronic methicillin-resistant Staphylococcus aureus (MRSA) osteomyelitis.
Materials and methods
The drug-loaded composite was developed using a solvent casting method and fabricated into four formulations: FV1, FV2, FV3, and FV4. The physicochemical characteristics and osteoblast biocompatibility were evaluated. In addition, the drug-release profile and anti-MRSA activity were examined.
Results
Vancomycin conserved its antibiotic property and did not form any interactions with the raw materials. A loading efficiency of more than 80 % was achieved with all formulations. FV2, FV3, and FV4 demonstrated cumulative release of more than 90 % over a couple of months. The bactericidal property was assessed to confirm the efficacy of the released vancomycin, and all composites showed antibacterial effects over 28 days. The release kinetics were best-fitted with the Korsmeyer–Peppas model, and Fickian diffusion was the main transport mechanism. A cytotoxicity test was also performed, and all formulations showed more than 80 % osteoblast viability.
Conclusions
All of the present in vitro results indicated the effectiveness of the new materials as local vancomycin carriers. In vivo animal models should be evaluated to confirm the efficacy for use in humans in the future.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chronic osteomyelitis is a major orthopedic problem and results in high morbidity and high cost of treatment [1]. Staphylococcus aureus is the most common causative pathogen for which beta-lactam antibiotics are the first-line options for treatment [2]. Long-term antibiotic administration for up to 4–6 weeks is required, and a resistant strain, methicillin-resistant Staphylococcus aureus (MRSA), has developed genetically. MRSA does not carry more intrinsic virulence than the original strains, but causes bone infections that are difficult to treat and can seriously damage the bone structure, leaving a poorly vascularized necrotic tissue (sequestrum) [3]. Vancomycin is the drug of choice. However, long-term administration of vancomycin results in renal toxicity. The concept of a local antibiotic delivery system has been proposed to directly carry the drug to the site of infection. By this means, a very high antibiotic concentration can be safely achieved at the infected site, while diminishing the systemic side effects [4].
Antibiotic-encapsulating polymethylmethacrylate (PMMA) is the gold standard for local osteomyelitis treatment. PMMA beads show slow and sustained release of the antibiotic without any toxicity toward bone cells. However, the beads are nonbiodegradable and a second operation is required for their removal [4, 5]. Many biodegradable carrier systems have been developed to solve this problem. The carriers can be fabricated from ceramics such as hydroxyapatite (HA) and β-tricalcium phosphate (β-TCP), or a combination of these two components as a biphasic calcium phosphate (BCP), and polymers such as poly(lactide)(PLA), poly(glycolide)(PGA), poly(ε-caprolactone)(PCL), and polyalkenoates (PHAs). The mechanical, biocompatibility, and resorbability properties can be finely adjusted and tuned by combining these components as composite materials. Such composite materials achieve a high modulus and strength from the ceramic phase, while remaining moldable because of the polymer phase [6].
For the ceramic part, BCP is chosen and the adjustable ratio of the slowly degraded HA and rapidly degraded β-TCP can serve a variety of medical applications including bone tissue engineering and local drug delivery systems [7]. For local drug carriers, different ratios of BCP have been used to encapsulate various antibiotics, such as doxycycline [8]. Besides, precipitation of the calcium and phosphate ions was found to induce osteoblast proliferation in both in vitro and in vivo experiments [9, 10]. The moldable property of the composite is obtained from the polymer phase. Poly(hydroxybutyrate-co-hydroxyvalerate)(PHBV), the most studied PHA, has been widely interested for medical applications. PHBV has both osteoblast biocompatibility and biodegradability and also possesses a piezoelectric characteristic resembling natural bone tissue [11]. Blending PHBV with other polymers, especially PCL was found to significantly improve its physicobiological properties as well as its drug release profile [12].
In this study, a PHBV/PCL blend was combined with BCP as a new composite material. The composite was used to encapsulate vancomycin to fabricate a new local antibiotic carrier for use in MRSA osteomyelitis. The physical and biocompatibility properties with osteoblasts were characterized. Finally, the vancomycin release profile and anti-MRSA activity were assessed.
Materials and Methods
Vancomycin-Loaded Composite Fabrication
PHBV (2.9 % 3-hydroxyvalerate) and PCL (MW 80,000) were purchased from Tianan Biologic Materials (Ningbo, China) and Shenzhen Bright China Industrial (Shenzhen, China), respectively. BCP was synthesized by the National Metal and Materials Technology Center (Pathumthani, Thailand). Briefly, pure HA and pure β-TCP powders in a 65:35 (w/w) ratio were mixed with ethanol for 22 h and dried overnight in an oven at 120 °C. A solvent casting method was performed to fabricate the vancomycin-loaded composite [13]. Briefly, PHBV and PCL blends in 20:80 and 30:70 (w/w) ratios were prepared by dissolving the polymers in chloroform at room temperature for 3 h. After homogeneous blends had been obtained, 20 and 30 % (w/w) BCP powder in relation to the polymer blends was added. The composite suspensions were stirred vigorously for 3 h at room temperature. Finally, vancomycin-loaded composites were prepared by adding vancomycin at a vancomycin-to-composite ratio of 1:10. After obtaining high viscosity, the suspensions were poured into glass plates creating thick films with a thickness of 5 cm each and subjected to chloroform evaporation for 48 h. A sharp circular razor of 1 cm in diameter was used to cut the evaporated materials film, and final products were labeled as FV1, FV2, FV3, and FV4 tablets (Table 1). Vancomycin-free tablets fabricated at each ratio were used as negative controls and labeled as F1, F2, F3, and F4.
Surface Topographic Assessment
Scanning electron microscopy (SEM) (Quanta 400; FEI, Hillsboro, OR) was performed for microscopic assessment of the surface topography of the drug-loaded composites. The tablets (FV1, FV2, FV3, and FV4) were attached to aluminum sample holders and coated with gold for 20 min. An acceleration voltage of 20 kV was used, and the surface topography was observed. SEM images of freshly prepared tablets and tablets after 56 days of immersion in phosphate-buffered saline (PBS) were examined.
Hardness Test
Hardness tests were performed using a hardness tester (PTB311E; Pharma Test, Hainburg, Germany), and reported in units of newtons at the time when each tablet was initially cracked (n = 5).
XRD Analysis
To examine the structural complexity and crystallinity of the vancomycin tablets, X-ray diffraction (XRD) analyses (X’Pert MPD; Phillips, Almelo, Netherlands) were performed on pure PHBV, PCL, BCP, vancomycin powder, and vancomycin tablets. Scanning angles of 2θ = 0–90° were used, and the XRD spectra were recorded and analyzed.
FTIR Analysis
The chemical interactions between vancomycin and composites were evaluated using Fourier transform infrared (FTIR) spectroscopy (Equinox 55; Bruker, Coventry, UK). For vancomycin-loaded composites and PCL film, the samples were cut into 1 × 2-cm pieces. For pure PHBV, BCP, and vancomycin powder, potassium bromide-containing pellets were prepared. All spectra were recorded in the 4000–400 cm−1 regions at room temperature. The peaks of the wavenumbers were compared among pure substrates and the drug-loaded composites.
Cytotoxicity of Vancomycin Tablets
Osteoblast Culture
MC3T3-E1 preosteoblast cells (lot no. 58633296; ATCC, Manassas, VA) at passages 4–10 were cultured in α-MEM (Gibco, Carlsbad, CA) supplemented with 10 % fetal bovine serum (FBS) (Gibco) and 100 μg/ml penicillin/streptomycin (Gibco). After reaching100 % confluency, the cells were trypsinized and seeded in 96-well plates at 5 × 103 cells/well. The cells were allowed to attach for 24 h at 37 °C in a 5 % CO2 atmosphere.
Vancomycin Tablet-Extract Preparation
Vancomycin tablets (n = 5 for each formulation) were sterilized by 25-kGy gamma-radiation. In accordance with ISO10993-12 for biological evaluation of medical devices, the samples were subjected to an indirect cytotoxicity (extraction) test [14]. Briefly, tablets were immersed in serum-free culture medium to achieve a surface area of 1.25 cm2/ml of medium for 24 h at 37 °C. The extract was then removed and mixed with FBS and serum-free culture medium in various ratios to give 0, 20, 40, 60, 80, and 100 % extract. The extract solutions were then added to previously prepared osteoblast cultures. The cells cultured with 0 % tablet extract (extract-free medium) were referred to negative controls. All cells were cultured for a further 48 h and then evaluated for their viability using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide), methyl thiotetrazole (MTT) assay.
Cell Viability Assay
Based on the activation of mitochondrial reductase enzymes in living cells, the MTT assay was quantitatively performed to assess the osteoblast viability [15]. Briefly, the culture medium was replaced with 0.5 mg/ml MTT (Merck, Whitehouse Station, NJ) solution, and the cells were incubated for 30 min. The purplish formazan crystals formed were dissolved in dimethylsulfoxide (Labscan, Samutsakorn, Thailand), and the resulting formazan solution was spectrophotometrically measured at 570 nm using a microplate reader (Spectromax M5; Molecular Devices, Sunnyvale, CA). The percentages of cell viability were obtained using the following formula:
where ODtreated and ODneg referred to the optical density (OD) of cells treated with extract-containing medium and cells cultured in extract-free medium, respectively. Dimethylsulfoxide was used as a blank solution, and its OD was represented as ODblank [16].
In Vitro Vancomycin Release Study
Determination of Drug-Loading Efficiency
A vancomycin standard curve was created using the OD at 280 nm of six dilutions of vancomycin from 5 to 2000 μg/ml. A linear regression equation for vancomycin concentration calculation was obtained. Individual tablets were weighed and immersed in 10 ml of chloroform (n = 5). After addition of an equal amount of distilled water, a rotating shaker apparatus was applied at 30 rpm for 2 h. Ultracentrifugation was then performed at 8000×g to separate the chloroform and water phases. Vancomycin dissolved in distilled water was carefully removed for OD measurement. The drug-loading efficiency was calculated using the following formula [17]:
Drug Release Profile Study
The drug release characteristics were evaluated by an in vitro elution method. Briefly, each tablet formulation (n = 5) was weighed and placed in 12-well plates containing 1.7 ml of PBS (pH 7.4) at 37 °C. After 6, 12, and 24 h, and 4, 8, 14, 21, 28, 35, 42, 49, and 56 days, the solution was collected and the OD was determined spectrophotometrically. An equal volume of fresh PBS was replaced at the predetermined time points. The release profile at each time point was assessed, and the cumulative release was also calculated. After 56 days of PBS immersion, the tablets were placed in a vacuum oven for 2 weeks and then weighed again to assess the percentages of weight loss using the following equation:
where W0 and Wt represent the initial dry weight and dry weight after a predetermined immersion time, respectively [18].
Release Kinetics Analysis
The drug release profiles of all formulations were fitted with mathematical models to determine the release kinetics and transport mechanisms. The cumulative data were fitted with various mathematical models including zero-order (F = k 0t), first-order (ln[1 − F] = −k 1t), Higuchi (F = k Ht1/2), and Korsmeyer–Peppas (F = k ptn) models. In all equations, F denotes the fraction of drug released at time t. The release constants for the zero-order, first-order, and Higuchi kinetic models were represented by k 0, k 1, and k H, respectively [19]. For the Korsmeyer–Peppas model, k p is the rate constant, while n is the diffusional exponent, which is used to indicate the drug transport mechanism. The best-fitted kinetic model for all formulations was then determined from the highest coefficient of determination (R 2) among the four models.
In Vitro Bactericidal Property of Composite-Loaded Vancomycin
To determine the antibacterial effects of the vancomycin-loaded tablets, a disc diffusion technique was used. The microbiological processes were conducted at the Microbiology Unit, Department of Pathology, Faculty of Medicine, Prince of Songkla University, Thailand. MRSA isolated from osteomyelitis patients in Songklanakarind Hospital, Thailand, was used in the experiment. Using conventional methods, pus specimens were initially inoculated on sheep blood agar, chocolate agar, and MacConkey agar plates. Biochemically, S aureus was identified using catalase and coagulase. Confirmation of the species was further performed using brain heart infusion broth. To select the resistant species, cefoxitin susceptibility was performed using a modified Kirby–Bauer disc diffusion method, in which an inhibition zone of less than 21 mm was considered to indicate resistance. The isolated MRSA was streaked on nutrient agar plates at a density of 5 × 107 CFU/cm3 and incubated for 24 h at 35 °C [20]. Subsequently, 6-mm antibiotic test discs soaked with the solutions from the drug release experiments were placed on the plates (three discs/plate). The plates were incubated for a further 18 h. The diameters of the inhibition zones were carefully measured using a Vernier digital caliper and assessed using the following criteria: zones of ≥12 mm were considered MRSA-sensitive; zones of 10–11 mm were considered intermediate; and zones of ≤9 mm were considered MRSA-resistant [21].
Statistical Analysis
For hardness tests, drug-loading efficiency determination, and vancomycin-tablet cytocompatibility, all data in each group (FV1, FV2, FV3, and FV4) were presented as means ± SD. Each variable was compared by one-way analysis of variance (ANOVA), and multiple comparisons between groups were performed using a Tukey post hoc test when a significant overall difference was detected. Statistical significance was set at p < 0.05 in a two-tailed test. For cumulative drug release, repeated-measures ANOVA was used to determine the significance of differences among groups, and values of p < 0.05 were considered significant.
Results
Physical Properties of the Vancomycin Tablets
Four formulations of vancomycin tablets prepared from PHBV, PCL, and BCP were successfully fabricated using a simple solvent casting method at room temperature (25–30 °C). The temperature changes during the fabrication processes downed to about 14 °C which may be due to the evaporation of chloroform in the reaction mixtures. Figure 1 shows the gross morphology of the vancomycin-loaded composites and their SEM images before and after PBS immersion. At day 0 (before PBS immersion), all of the tablets showed scattered microporosity. After 56 days of immersion, the microporosity was further distributed and obviously increased throughout the tablets compared with the initial images taken before the incubation.

Gross morphology of the vancomycin tablets and their SEM images. a, d, g, j The gross morphology of FV1, FV2, FV3, and FV4, respectively. b, e, h, k The surface topography at day 0, respectively. c, f, i, l The surface topography at day 56, respectively
The physical properties of the vancomycin tablets were examined, and their components, hardnesses, and weights are summarized in Table 1. The FV1 formulations showed the highest hardness and lowest weight loss percentage, and the differences were significant. The highest loading efficiency also belonged to FV1 tablets. However, the loading efficiencies did not differ significantly among the formulations and more than 80 % of vancomycin could be encapsulated in all types of composites.
Chemical Property
XRD analyses were performed to assess the crystallinity of the vancomycin-loaded composites, as illustrated in Fig. 2. After the addition of vancomycin, all formulations showed crystallinity. The characteristic peaks of the pure substances were not disturbed and were clearly observed in all formulations. Notably, vancomycin showed an amorphous characteristic illustrated by its fingerprint-appearance spectrum, in which no specific peaks could be identified. Compared with the spectra of vancomycin-free composites (Fig. 3), the drug-loaded tablets showed the same crystallinity patterns.

XRD spectra of the raw materials and vancomycin-loaded composites

XRD spectra of the vancomycin-free composites
The FTIR spectra of all vancomycin tablets are shown in Fig. 4. Both PHBV and PCL, which are aliphatic polyesters, typically showed a C=O stretching band at 1724 cm−1. The symmetric –C–O–C– groups were indicated by wavenumbers at 800–975 cm−1, whereas the stretching bands located at 1060–1150 cm−1 were antisymmetric –C–O–C– groups [12]. The most prominent polyester characteristic band located at 1724 cm−1 was conserved in all vancomycin-loaded composites. Regarding the BCP spectrum, two intense PO4 3− bands were located at 1074 and 1033 cm−1, and these bands were still conserved in the drug-loaded tablets. Compared with the FTIR spectra of the drug-free composites (Fig. 5), the drug-loaded spectra did not show any significant differences. For the vancomycin spectrum, the absorption bands were typically found at 3450, 1651, 1504, and 1230 cm−1 for hydroxyl stretching, C=O stretching, C=C, and phenols, respectively [22]. These bands were not clearly found in the drug-loaded spectra of composites, since only about 10 % of the drug was in the formulations. However, the release profiles (Fig. 6) illustrating more than 90 % of cumulative releases indicated that the drug did not form covalent bond with the main ingredients in the composites. Possible interaction in some formulation (FV1) could be observed due to hydrogen bonding interaction resulting in the slowest release pattern.

FTIR spectra of the raw materials and vancomycin-loaded composites

FTIR spectra of the vancomycin-free composites

Cumulative release of vancomycin from the drug-loaded tablets
Drug Release Study
The percentage of vancomycin released from all tablets was normalized to the total amount of vancomycin incorporated into the composites. The cumulative data are shown in Fig. 6, and the concentrations of released vancomycin in each time period are illustrated in Fig. 7. During the first 24 h, all formulations exhibited rapid drug release, in which FV4 showed the highest accumulative dissolution of 41.98 %. Overall, after 1 day of immersion, the release rates decreased and the curves changed their slopes. At the end of experiment, more that 90 % of vancomycin was released from FV2, FV3, and FV4, while only 50.12 % of vancomycin was released from FV1.

Concentrations of vancomycin released in relation to the time points
Regarding the release profile in each time period (Fig. 7), although the release rate was significantly decreased after 1 week, almost all of the vancomycin concentrations remained higher than the minimum inhibitory concentration (MIC) of 2 μg/ml for S. aureus [23]. The initial burst release showed a concentration of more than 1000 μg/ml, and the highest amounts of vancomycin release were found at the fourth day for all formulations. The concentrations decreased dramatically with minimal fluctuation overtime.
Drug Release Kinetics
Table 2 summarizes the release constants, coefficients of determination (R 2), and release exponents (n) for the Korsmeyer–Peppas model. According to the mathematic model fitting, the coefficient of determination (R 2) for the Korsmeyer–Peppas model was highest in all formulations. For this reason, the best-fitted kinetics for all carriers were assigned to the Korsmeyer–Peppas model. Regarding the n value, all of the formulations showed n values of less than 0.45. According to the model, the k p value was used to determine the release rate, and FV4 showed the highest rate (32.37), while FV1 exhibited the lowest rate (13.94).
In Vitro Bactericidal Property of Vancomycin
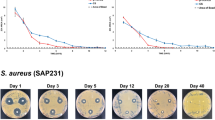
To confirm the vancomycin activity, a disc diffusion test was performed. The inhibition zones in relation to the drug concentration of each formulation are shown in Fig. 8. MRSA sensitivity (zones larger than 12 mm) was found for most of the tablets until 1 month of immersion. At 35 days, FV1, FV2, and FV4 showed moderate susceptibility, while FV3 still exhibited MRSA sensitivity. However, from day 42, all formulations showed decreased susceptibility and finally changed to MRSA resistance at the end of the experiment.

Inhibition zones in relation to vancomycin concentrations. a, b, c, d The results for FV1, FV2, FV3, and FV4, respectively
Indirect Cytotoxicity Test of Vancomycin Tablets
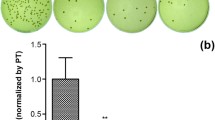
An indirect (extract) cytotoxicity test was performed to confirm that vancomycin released from carriers did not cause unacceptable cell death. Based on the rapid drug release in the first 24 h, extracts were prepared by immersion of the tablets for 24 h. The extracts at various dilutions were then applied to osteoblast cultures. After a further 48 h of culture, MTT assays were performed. The results are shown in Fig. 9. Osteoblasts cultured with all tablet extracts demonstrated more than 80 % cell survival even in the 100 % extract condition.

Indirect cytotoxicity test of the vancomycin tablets using the MTT assay
Discussion
Osteomyelitis creates both infectious and bone defect problems. The vancomycin-loaded composites represented the novel local antibiotic carrying systems managing the infected dead space. The antibiotic was slowly released from the composites and eradicated the microorganisms while maintaining the space for new bone formation. The new bone will theoretically form replacing the degraded composites. In this study, the vancomycin-loaded PHBV/PCL and BCP composites were successfully fabricated into four formulations, FV1, FV2, FV3, and FV4, using a conventional solvent casting method. No exothermic reactions were observed throughout the fabrication processes, which could suggest a potency for encapsulating heat-labile molecules without degradation or denaturation. However, if the evaporation process was incomplete, the organic solvent would remain as a contaminant and cause cell damage [24].
Overall, the physical properties appeared to be similar among the four vancomycin-loaded composites, yet their hardnesses were quite different. FV1 showed the highest hardness, while FV4 exhibited the lowest hardness. This could arise because FV4 contained the lowest ratio of the plasticizer PCL, which may not be able to hold the integrity after addition of vancomycin. This result was consistent with the maximum weight loss observed in FV4. These phenomena support the theory that as the filler (ceramic) loading increases, more mass loss will occur in the material [25]. Generally, higher amounts of antibiotics impregnated into carriers have been known to decrease the mechanical properties, as seen in vancomycin-incorporated PMMA [26]. Too much encapsulated drug may result in an incapability to mold the composite/polymer into tablets or other shapes. This might also lead to cracking and excessive drug release into the local environment. Clinically, the antibiotic-loaded system cannot maintain a large infected tissue defect without any other fixation system. The drug-loaded composite only acted as a temporary spacer, and the ability for maintaining its dimensional integrity during the release process is the key concept.
Theoretically, the amount of drug loading should be nearly equal to the real amount in the tablet, and the manufacturing processes should preserve the initial loading as much as possible. In our experiments, the loading efficiencies were more than 80 % for all formulations, suggesting highly effective fabrication processes. In other words, nearly all of the loaded drug had a chance to be released and kill bacteria.
Chemically, the same FTIR and XRD spectra were observed in the drug-loaded composites compared with the vancomycin-free samples. These findings suggest that addition of vancomycin did not lead to chemical interactions with any of the PHBV, PCL, or BCP moieties. Regarding vancomycin, the water-soluble form was used in these experiments because it mimics real clinical practice, in which the water-soluble form is preferably used over the pure crystallized compound. As a result, crystallinity peaks were not detected, and the amorphous pattern remained even when the drug had been encapsulated.
The drug release studies were performed for 56 days, which reflects the duration required for bacterial eradication and new bone regeneration [3, 5]. All of the formulations showed slow release for over 2 months, and nearly 100 % of cumulative releases were documented from FV2, FV3, and FV4. These results suggest the potential of these formulations for use as vancomycin vehicles for the eradication of MRSA. Regarding the lowest release in FV1, strong physical bonding between the drug and the materials supported by the highest hardness might have contributed to the difficulty in diffusion of the drug from the carrier.
The release patterns were initiated with a rapid release phase occurring through dissolution of the drug located on the surface of the carriers [22]. This rapid elution has an advantage, in that a high concentration of released antibiotic can penetrate biofilms, indicating an increased chance for complete eradication of the pathogen [27]. However, cell death was reported when the local level of vancomycin reached 5000–10,000 μg/ml, comprising 2–5-fold greater levels than the vancomycin released from the composites [26]. From the extract test, more than 80 % cell viability was observed in all batches, indicating a very low tendency for cytotoxicity toward bone cells according to ISO 10993–12 (part 5) [28]. In the second stage, the dissolution rate declined, as demonstrated by the changing slopes, and the carriers then exhibited a long release period over a couple of months. This phase basically occurred through buffer diffusion into the swelling carrier through micropores, which resulted from the fabrication processes and/or the possible footprint left by the surface-located vancomycin molecules after dissolution. The dissolved drug then diffused into the surrounding buffer according to the concentration gradient through those pores [29]. The last phase exhibited a plateau pattern in which the very low amount of remaining drug was finally diffused. Although this is the first study to propose a novel PHBV, PCL, and BCP composite as a vancomycin carrier, different kind of materials have previously been used for vancomycin delivery experimentally. Yang et al. fabricated a chitosan/HA composite, and the material reached approximately 99 % of cumulative release during 32 days of incubation [30]. Li et al. applied a PHBV-coated bioglass for vancomycin release and found a good cumulative release of 95 %, but the elution was only sustained for 10 days [31].
In the kinetic study, the release data were fitted with four mathematic models to clarify the most appropriate release kinetics. All of the vancomycin-incorporated composites showed the highest coefficient of determination (R 2) after fitting with the Korsmeyer–Peppas model. This model describes the drug release from a polymeric system and was strongly correlated with the carriers because they were mainly fabricated from polymeric materials, PHBV and PCL [32]. The release constant (k p) is used to determine the release rate. FV4 exhibited the highest rate, because of its lowest physical bonding with the carrier. Focusing on the release exponent (n), this value is an indicator of the release mechanism and depends on the matrix carrier geometry. All of the vancomycin tablets demonstrated n values of less than 0.45. Fickian diffusion is the main drug transport mechanism under the conditions used [33]. Although all of the formulations showed effective release over a couple of months, the bactericidal property of the dissolved vancomycin also required confirmation. The inhibition zones were correlated with the concentrations released. Interestingly, after 28 days of immersion, the zone diameter decreased to less than 10 mm, interpreted as vancomycin resistance, while the concentration of released vancomycin remained above the MIC. This might be an example of a disc diffusion test limitation. The interpretation of the test, namely the diameter of the inhibition zone, is mainly affected by the concentration gradient of the drug and the agar. In the late phase of the release, the drug concentration may have been too low for diffusion. In addition, a high-molecular-weight antibiotic such as vancomycin may diffuse from the disc into the agar with greater difficultly than a smaller molecule. However, the bactericidal period of 28 days was acceptable, because it was corresponded to the osteomyelitis treatment course lasting approximately 4–6 weeks [3, 5].
Conclusions
In this study, a novel local vancomycin delivery system was produced. Four vancomycin-loaded composites were successfully fabricated, and loading efficiencies of more than 80 % were achieved in all formulations. All of the carriers exhibited slow and sustained release of the antibiotic over 56 days with retained bactericidal property for 4 weeks. More than 90 % of vancomycin was released from FV2, FV3, and FV4. All of the release kinetics were fitted with the Korsmeyer–Peppas model, and Fickian diffusion was the transport mechanism. An indirect cytotoxic test was performed to examine whether the tablet extracts could have detrimental effects on cells, and cell proliferation rates of more than 80 % were confirmed, even at the highest extract concentration. In conclusion, this new vancomycin delivery system showed positive tendencies for use as an antibiotic carrier in the treatment of chronic MRSA osteomyelitis. Further in vivo animal studies should be performed to confirm the efficacies for clinical use in humans.
Abbreviations
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- PMMA:
-
Polymethylmethacrylate
- HA:
-
Hydroxyapatite
- β-TCP:
-
β-Tricalcium phosphate
- BCP:
-
Biphasic calcium phosphate
- PLA:
-
Poly(lactide)
- PGA:
-
Poly(glycolide)
- PCL:
-
Poly(ε-caprolactone)
- PHAs:
-
Polyhydroxyalkanoates
- PHBV:
-
Poly(hydroxybutyrate-co-hydroxyvalerate)
- SEM:
-
Scanning electron microscopy
- XRD:
-
X-ray diffraction pattern
- FTIR:
-
Fourier transformed infrared
- MTT:
-
(3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide), methyl thiotetrazole
References
Calhoun JH, Manring MM, Shirtliff M. Osteomyelitis of the long bones. Semin Plast Surg. 2009;23:59–72.
Rao N, Ziran BH, Lipsky BA. Treating osteomyelitis: antibiotics and surgery. Plast Reconstr Surg. 2011;127:177–87.
Lazzarini L, Mader JT, Calhoun JH. Osteomyelitis in long bones. J Bone Joint Surg Am. 2004;86:2305–18.
Silva GA, Ducheyne P, Reis RL. Materials in particulate form for tissue engineering. 1. Basic concepts. J Tissue Eng Regen Med. 2007;1:4–24.
Xie Z, Liu X, Jia W, Zhang C, Huang W, Wang J. Treatment of osteomyelitis and repair of bone defect by degradable bioactive borate glass releasing vancomycin. J Control Release. 2009;139:118–26.
Causa F, Netti PA, Ambrosio L, Ciapetti G, Baldini N, Pagani S, et al. Poly-epsilon-caprolactone/hydroxyapatite composites for bone regeneration: in vitro characterization and human osteoblast response. J Biomed Mater Res A. 2006;76:151–62.
Kumar TSS, Manjubala I, Gunasekaran J. Synthesis of carbonated calcium phosphate ceramics using microwave irradiation. Biomaterials. 2000;21:1623–9.
Victor SP, Kumar TS. BCP ceramic microspheres as drug delivery carriers: synthesis, characterisation and doxycycline release. J Mater Sci Mater Med. 2008;19:283–90.
Fellah BH, Gauthier O, Weiss P, Chappard D, Layrolle P. Osteogenicity of biphasic calcium phosphate ceramics and bone autograft in a goat model. Biomaterials. 2008;29:1177–88.
Zhu XD, Fan HS, Xiao YM, Li DX, Zhang HJ, Luxbacher T, et al. Effect of surface structure on protein adsorption to biphasic calcium-phosphate ceramics in vitro and in vivo. Acta Biomater. 2009;5:1311–8.
Freier T. Biopolyesters in tissue engineering applications. Adv Polym Sci. 2006;203:1–61.
Mendes JB, Riekes MK, De Oliveira VM, Michel MD, Stulzer HK, Khalil NM, et al. PHBV/PCL microparticles for controlled release of resveratrol: physicochemical characterization, antioxidant potential, and effect on hemolysis of human erythrocytes. ScientificWorldJournal. 2012;2012:1–13.
Mehrabanian M, Nasr-Esfahani M. HA/nylon 6,6 porous scaffolds fabricated by salt-leaching/solvent casting technique: effect of nano-sized filler content on scaffold properties. Int J Nanomedicine. 2011;6:1651–9.
Sarasua JR, Lopez-Rodriguez N, Zuza E, Petisco S, Castro B, Del Olmo M, et al. Crystallinity assessment and in vitro cytotoxicity of polylactide scaffolds for biomedical applications. J Mater Sci Mater Med. 2011;22:2513–23.
Cetin D, Kahraman AS, Gümüşderelioğlu M. Novel pHEMA-gelatin SPHs as bone scaffolds in dynamic cultures. J Mater Sci Mater Med. 2012;23:2803–12.
Jiang T, Li Y, Lv Y, Cheng Y, He F, Zhuo R. Biodegradable amphiphilic block-graft copolymers based on methoxy poly(ethylene glycol)-b-(polycarbonates-g-polycarbonates) for controlled release of doxorubicin. J Mater Sci Mater Med. 2013;25:131–9.
Mundargi RC, Srirangarajan S, Agnihotri SA, Patil SA, Ravindra S, Setty SB, et al. Development and evaluation of novel biodegradable microspheres based on poly(d, l-lactide-co-glycolide) and poly(epsilon-caprolactone) for controlled delivery of doxycycline in the treatment of human periodontal pocket: in vitro and in vivo studies. J Control Release. 2007;119:59–68.
Wang Q, Wang Q, Wan C. The effect of porosity on the structure and properties of calcium polyphosphate bioceramics. Ceramics. 2011;55:43–8.
Ranjha NM, Ayub G, Naseem S, Ansari MT. Preparation and characterization of hybrid pH-sensitive hydrogels of chitosan-co-acrylic acid for controlled release of verapamil. J Mater Sci Mater Med. 2010;21:2805–16.
Lin SS, Ueng SW, Liu SJ, Chan EC, Chao EK, Tsai CH, et al. Development of a biodegradable antibiotic delivery system. Clin Orthop Relat Res. 1999;362:240–50.
Kankilic B, Bayramli E, Kilic E, Dağdeviren S, Korkusuz F. Vancomycin containing PLLA/beta-TCP controls MRSA in vitro. Clin Orthop Relat Res. 2011;469:3222–8.
Yao Q, Nooeaid P, Roether JA, Dong Y, Zhang Q, Boccaccini AR. Bioglass®-based scaffolds incorporating polycaprolactone and chitosan coatings for controlled vancomycin delivery. Ceram Int. 2013;39:7517–22.
Canut A, Isla A, Betriu C, Gascóm AR. Pharmacokinetic-pharmacodynamic evaluation of daptomycin, tigecycline, and linezolid versus vancomycin for the treatment of MRSA infections in four western European countries. Eur J Clin Microbiol Infect Dis. 2012;31:2227–35.
Dorozhkin SV. Biocomposites and hybrid biomaterials based on calcium orthophosphates. Biomatter. 2011;1:3–56.
Tang CY, Chen DZ, Yue TM, Chan KC, Tsui CP, Yu PHF. Water absorption and solubility of PHBHV/HA nanocomposites. Compos Sci Technol. 2008;68:1927–34.
Urabe K, Naruse K, Hattori H, Hirano M, Uchida K, Onuma K, et al. In vitro comparison of elution characteristics of vancomycin from calcium phosphate cement and polymethylmethacrylate. J Orthop Sci. 2009;14:784–93.
Rossi S, Azghani AO, Omri A. Antimicrobial efficacy of a new antibiotic-loaded poly(hydroxybutyric-co-hydroxyvaleric acid) controlled release system. J Antimicrob Chemother. 2004;54:1013–8.
International Organization for Standardization. Biological evaluation of medical devices—Part 5: Tests for in vitro cytotoxicity (ISO 10993–5:2009). Geneva: ISO; 2009. p. 1–34.
Ning Z, Xiongbiao C. Biofabrication of tissue scaffolds. In: Pignatello R, editor. Advances in biomaterials science and biomedical applications. Rijeka: InTech; 2013. p. 315–28.
Yang CC, Lin CC, Liao JW, Yen SK. Vancomycin-chitosan composite deposited on post porous hydroxyapatite coated Ti5Al4V implant for drug controlled release. Mater Sci Eng C Mater Biol Appl. 2013;33:2203–12.
Li W, Nooeaid P, Roether JA, Schubert DW, Boccaccini AR. Preparation and characterization of vancomycin releasing PHBV coated 45S5 Bioglass®-based glass-ceramic scaffolds for bone tissue engineering. J Eur Ceram Soc. 2014;34:505–14.
Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35.
Soundrapandian C, Datta S, Kundu B, Basu D, Sa B. Porous bioactive glass scaffolds for local drug delivery in osteomyelitis: development and in vitro characterization. AAPS PharmSciTech. 2010;11:1675–83.
Acknowledgments
This work was supported by the Faculty of Medicine, Prince of Songkla University, Songkhla, Thailand, under grant EC 522821142. The authors would like to thank Dr. Naruporn Monmaturapot from MTEC, Thailand, for the BCP syntheses and Assoc. Prof. Dr. Chitchamai Ovartlanporn, Department of Pharmaceutical Chemistry, Faculty of Pharmaceutical Sciences, Prince of Songkla University, Thailand, for the chemical analyses.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Upho, N., Tangtrakulwanich, B., Pripatnanont, P. et al. Development of Novel PHBV/PCL and BCP Composite for Musculoskeletal Infection: an In Vitro Vancomycin Release and Anti-MRSA Effect. J Pharm Innov 10, 211–221 (2015). https://doi.org/10.1007/s12247-015-9219-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12247-015-9219-4