
Abstract
Pollen protoplasts provide a sexual and haploid system for haploid production, cell fusion and mutation studies used in plant improvement. Due to the multiploidy, heterozygosity, and often self-incompatibility in tetraploid genotypes, haploid potatoes are desirable for breeding schemes via ploidy manipulations. In this study, two tetraploid varieties and two dihaploid lines of potato were used for pollen tetrad protoplast isolation and culture. The meiotic tetrad buds were first pre-treated at 5 °C for 0–12 days, then the tetrads were transferred into enzyme solutions containing different concentrations of snailase (0.5–1.5%), 0.3 M osmolites (sucrose, mannitol, glucose or sorbitol), 1.0% Cellulose, 0.5% Hemicellulase, 0.5% Pectolyase, 0.3% Sucrose, 3 mM 2-(N-Morpholino) ethane sulfonic acid, 1% polyvinyl pyrrolidone, 0.01% casein hydrolysate and K3 medium compositions. Among the four donor materials, tetraploid cv. Gannongshu No. 3 (‘GNS No.3’) showed the greatest protoplast yield (74.6 ± 2.4%). In this variety, most of the tetrad protoplasts regenerated a cell wall and continued cell divisions were observed when they were inoculated in K3 basic medium supplemented with (0.5–1.0) mg/L 2,4-D + (0.1–0.5) mg/L KT + 0.4 mg/L 6-BA +800 mg/L glutamine +100 mg/L serine. ‘GNS No.3’ also showed the greatest first division frequency (21.6 ± 1.5%) and sustained division to form multicellular structures. The study findings suggested that cultured tetrad pollen protoplasts could reverse the gametophytic developmental pattern programmed in vivo to a sporophytic pathway leading to multicellular microspore-derived colonies.
Resumen
Los protoplástos del polen suministran un sistema sexual y haploide para la producción de haploides, fusión de células y estudios de mutación que se usan en el mejoramiento de plantas. Debido a la multiploidía, heterozigocidad, y a menudo la autoincompatibilidad en genotipos tetraploides, se desean las papas haploides para esquemas de mejoramiento por vía de manipulaciones de la ploidía. En este estudio, se usaron dos variedades tetraploides y dos líneas dihaploides de papa para aislamiento y cultivo de protoplastos de polen en estado de tétradas. Las yemas de tétradas meióticas se pre-trataron primero a 5 °C de 0 a 12 días, después se transfirieron las tétradas a soluciones de enzimas que contenían diferentes concentraciones de snailasa (0.5–1.5%), 0.3 M de osmolitos (sacarosa, manitol, glucosa o sorbitol), 1.0% de celulasa, 0.5% de hemicelulasa, 0.5% de pectoliasa, 0.3% de sacarosa, 3 mM de ácido etano-sulfónico 2-(N-morfolina), 1% de polivinil-pirrolidona, 0.01% de hidrolizado de caseína, y composiciones de medio K3. Entre los cuatro materiales donantes, la variedad tetraploide Gannongshu No. 3 (‘GNS No.3’) mostró el más alto rendimiento en protoplástos (74.6 ± 2.4%). En esta variedad, la mayoría de los protoplastos de tétradas regeneraron una pared celular, y se observaron continuas divisiones celulares cuando se inocularon en medio básico K3 suplementado con (0.5–1.0) mg/L 2,4-D + (0.1–0.5) mg/L KT + 0.4 mg/L 6-BA +800 mg/L glutamina +100 mg/L serina. “GNS No. 3” también mostró la mayor frecuencia de la primera division (21.6 ± 1.5%) y división sostenida para formar estructuras multicelulares. Lo que se encontró en este estudio sugiere que protoplastos cultivados de polen en tétradas pudieran revertir el patrón de desarrollo gametofítico programado in vivo, a una ruta esporofítica que conduzca a colonias multicelulares derivadas de microsporas.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant protoplasts are cells from which the cell wall has been enzymatically removed. Protoplasts are attractive model organisms for studies of physiological and cytological processes related to cell wall formation, cell growth, and differentiation being used in basic research and plant improvement (Fan et al. 2001; Yoo et al. 2007). In comparison with somatic protoplasts, pollen protoplasts provide an original sexual and haploid system for haploid induction, cell fusion, genetic manipulation, mutation studies and they are used in plant improvement, as well as for studying pollen biology (Bajaj and Davey 1974; Lee and Power 1988; Desprez et al. 1995; Fang et al. 2006). In young microspore tetrads of some angiosperms, each spore is initially one of four haploid cells enclosed in the callose wall (Bhojwani and Cocking 1972). The initial wall material is cellulosic and is followed by the deposition of lipoprotein and, finally sporopollenin which is enzyme-resistant (Bhojwani and Cocking 1972; Loewus et al. 1985). Because the callose wall can be degraded by snail enzyme with high β-1,3 glucanase activity, pollen tetrad protoplasts have been successfully isolated from some angiosperms like Datura metel (Rajasekhar 1973), Atropa belladonna, Nicotiana tabacum and Triticum aestivum (Bhojwani and Cocking 1972; Bajaj 1974), Cajanus cajan and Zea mays (Deka et al. 1977), and Digitalis obscura (Arnalte et al. 1991). A sufficient population of the isolated protoplasts must remain viable to be used in downsteam procedures, such as culturing, fusion or transformation. Although culture of pollen protoplasts was pioneered by Bajaj and Davey (1974), not much progress has been made due to technical difficulties.
The cultivated potato (Solanum tuberosum L.), which is a tetraploid (2n = 4× = 48), is ranked as the third most important global food crop and the most widely grown noncereal crop (Hijmans 2001; Barrell et al. 2013). Potato tubers are a globally important dietary source of starch, protein, antioxidants and vitamins, serving the plant as both a storage organ and a vegetative propagation system (Burlingame et al. 2009; Barrell et al. 2013). S. tuberosum genotypes are generally heterozygosous, occur in a range of ploidies, and are often self-incompatible at the diploid level, causing many problems such as relatively long breeding cycles and inbreeding depression (Wenzel et al. 1979; Conner et al. 1997; Muthoni et al. 2015). Due to the complex nature of genetic interactions among genes in tetraploid genotypes, dihaploid potatoes are desirable for the study of genetic interactions in a less complex background (Wenzel et al. 1979; Ortiz 1998). Although anther and isolated microspore cultures have been developed to produce haploid and doubled-haploid plants (Germanà 2011; Ferrie and Caswell 2011), these methods have not been effective for many genotypes and the number of haploids obtained remain low in all the cases despite considerable efforts. It suggests that alternative methods should be developed to produce haploids in recalcitrant genotypes and to improve induction efficiency of genotypes already producing haploids (Germanà and Chiancone 2001). If protoplasts could be isolated from pollen tetrads they can serve as a starting material for the production of haploid plants as well as for fusion studies in relation to somatic hybridization of potato. However, compared to other angiosperms, the isolation methods for protoplasts from potato pollen is a pending research issue. In spite of numerous studies reporting successful protoplast isolation from somatic cells of domestic cultivars and various species of potato, their subsequent culture and regeneration into plants (Tavazza and Ancora 1986; Dai et al. 1987; Jones et al. 1989), there is not a satisfactory method for isolation of abundant pollen protoplasts from potato.
Based on previous studies about isolation of mature pollen protoplasts from potato (Zhang et al. 2004), the purpose of this study was to develop a reproducible method for the isolation of a large population of viable tetrad protoplasts. In the present study, a protocol was developed for pollen tetrad protoplast isolation from two tetraploid local cultivars (2n = 4× = 48) and two dihaploid lines (2n = 2× = 24) and some observations on the culture behavior of these pollen tetrad protoplasts were reported.
Materials and Methods
Plant Materials
Two tetraploid (Solanum tuberosum) varieties of local cultivars named ‘Gannongshu No. 1’ (GNS No.1) (2n = 4× = 48), ‘Gannongshu No. 3’ (GNS No.3) (2n = 4× = 48) and two dihaploid lines of ‘81–15’ (2n = 2X = 24) and ‘81–8’ (2n = 2× = 24) obtained by in vitro androgenesis were grown in an experimental field in Qin Wangchuan (103°31′17.22″ E, 36°27′18.29″ N, 1950 m.a.s.l.), Gansu Province, Northwest region of China. The plants were planted in late April in 7 × 7 m2 plots replicated three times in a randomized complete block design. Potatoes were spaced at a distance of 0.30 m in row spacing and 0.80 m between rows. Distance between plots and blocks was 0.80 and 1.2 m, respectively. Young flower buds (close to tetrad stage) and mature pollen grains (from undehisced anthers of just opening flowers) were collected on a sunny day at 10:00–11:00 am in July and preserved at 4 °C.
Isolation of Protoplasts
After the young flower bud collection, only buds containing anthers with tetrads were selected. For this aim, one of the five anthers of a bud was removed and stained with acetocarmine, then observed under a microscope (10 × 10) to examine the stage of spore development. If the squashed anther contained pollen tetrads, then the bud was retained. Selected buds were pre-treated at 5 °C at different durations (0, 3, 7, 10 or 12 days), then successively surface-sterilized with 70% (v/v) ethanol for 30 s, then soaked for 5–7 min in 0.1% HgCl2 (w/v) and rinsed 5–6 times with sterile double distilled water. The basal end of each anthers was cut under water and pollen tetrads were squeezed out using a scapel, then the supernatants were filtered through a steel sieve (150 μm pore size) to remove surplus anther wall debris. The filtrate was centrifuged three times at 500 rpm for 5 min and the supernatants were poured into 10 ml of filter-sterilized enzymatic mixture containing [0.5, 0.8, 1.0, 1.2 and 1.5% (w/v)] snailase (Sigma-Aldrich, St. Louis, Mo, USA), 1% (w/v) cellulose (Onozuka R-10, Yakult Co. Ltd., Tokyo, Japan), 0.5% (w/v) hemicellulase (Sigma H-2125, Aldrich, St. Louis, Mo, USA), 0.5% (w/v) pectinase (Sigma-Aldrich, St. Louis, Mo, USA), 3 mM MES (Sigma-Aldrich, St. Louis, Mo, USA), 0.01% CH (Sigma-Aldrich, St. Louis, Mo, USA), 1% PVP (MW 10,000, Sigma-Aldrich, St. Louis, Mo, USA) dissolved in K3 medium (Kao et al. 1974). The enzyme mixture also contained 0.3 M of (sucrose, mannitol, glucose or sorbitol) (as osmoticum) at pH 5.6 and incubated in the dark at 24–26 °C with gentle shaking (60 rpm) for 2–4 h. After 4 h, when most pollen tetrad protoplasts had been released, the pollen suspension was centrifugated and the supernatant was removed. Then the sedimentation was washed and purified by discontinuous centrifugation twice at 500 rpm for 10 min followed by floatation on K3 medium containing 25% sucrose and 50 mM MES, pH 5.6. The floated protoplasts were collected using a pipette.
Fluorescein diacetate (FDA, Sigma) was dissolved in acetone to produce l mg/mL stock solution. For viability tests, the FDA stock was added to the medium to final concentration of 1 mg/mL. To determine whether the cell wall was present or absent on the surface of the isolated pollen tetrad protoplasts, we applied a solution of 0.1% Calcofluor White ST (CW, Sigma) dissolved in 0.5 M sucrose. After 10 min of staining, the protoplasts were collected and transferred into dye-free medium.
Culture of Protoplasts
Isolated protoplasts were suspended into a 30 mm diam sterile petri plate at a density of 1 × 103–104 protoplasts per mL in a modified K3 medium lacking NH4NO3 and NAA, and containing organic additives: 800 mg/L glutamine, 100 mg/L serine, 0.3 M sucrose, 0.1% calf serum and one of eight plant growth regulators combinations (Table 1) with pH at 5.6. The medium was filter-sterilized. The petri plates were then sealed with Parafilm and incubated at 28 ± 2 °C in the dark and observed every day. When a regenerated cell finished the first division, the petri plates were moved to low light (≤ 20 μE m−2 s−1). After 7–10 days the cultures were diluted with fresh medium. The cultures were incubated with gentle shaking (60 rpm) for 2–3 min per 6 h every day at 28 °C at beginning and after that they were maintained in stationary conditions.
Observations and Data Analysis
The isolation and culture of protoplasts were observed under an Olympus IMT-F-4 inverted fluorescence microscope, with UV illumination and an excitation filter 420 nm and a blocking filter 530 nm. The data presented are mean values in ten randomly selected microscopic fields. Data were subjected to one-way analysis of variance (ANOVA) and Duncan’s multiple range tests for each parameter at P < 0.05 using SPSS13.0. The percentage of full mature pollen grains (PFPG), protoplast isolation rate (PIR) and division frequency (DF) in percentages were calculated as follows:
Results
Effect of Genotype and Ploidy Level on Protoplast Isolation
Protoplast yields strongly depended on the genotype and ploidy level of the donor plant (Table 2). Tetraploid varieties (GNS No.1, GNS No.3) showed significant greater numbers of protoplasts (68.5–74.6%) and division frequency (18.4–21.6%) than haploid lines (Table 2). In contrast, few protoplasts (2.3–43.8%) were released in haploid lines and hardly any division was observed from ‘81–8’ line. With equivalent percentages of full mature pollen grains between GNS No. 1 and GNS No. 3, but improved isolation rate of GNS No. 3, we selected GNS No. 3 for subsequent experiments on tetrad protoplast isolation.
Effect of Snailase Concentration and Osmotic Stabilizer on Protoplast Isolation
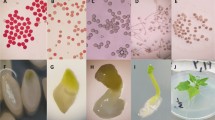
More intact protoplasts and less budding protoplasts were obtained when sucrose was used as osmotic stabilizer in comparison to the other three tested stabilizers. Protoplast release was attained at with 0.3 M sucrose at all concentrations of snailase >0.5%; however, at 1.5% snailase and 0.3 M sucrose, we observed protoplast breaking and budding (Table 3). The enzymatic solution consisting of 1.2% snailase and 0.3 M sucrose as osmotic pressure regulator showed high protoplast isolation rates and yielded more intact and dividing protoplasts, so it was used in the following experiments. Isolated pollen tetrad protoplasts were spherical, with dense cytoplasm, a visible nucleus which occupied a large portion of the protoplast, and lacked cell wall remnants (Fig. 1a-c). Strong FDA fluorescence and active cytoplasmic streaming observed by microscopy confirmed the viability of the majority of the pollen tetrad protoplasts (Fig. 1d).

Isolation and culture of pollen tetrad protoplasts from Solanum tuberosum. a. Freshly collected tetrad microspores (10 × 10). Bar = 20 μm; b-c. Population of pollen tetrad protoplasts (10 × 40). Bar = 20 μm; d. Viability of pollen tetrad protoplasts tested with fluorochromatic reaction with FDA (10 × 10); e-f. Two-celled units produced by first division from regenerated cell after 3–4 days of culture (10 × 40). Bar = 20 μm; g. Secondary division of regenerated cell after 6–8 days of culture (10 × 10); h. A multicellular cluster after 20 days of culture (10 × 10)
Effect of Cold Pre-Treatment on Protoplasts Isolation and Culture
The highest protoplast yield (73.1 ± 2.4%) and viability (95.2 ± 2.9%) and division frequency (21.6 ± 1.2%) were obtained when the buds were pre-treated with low temperature (5 °C) for 7 days (Table 4). This treatment allowed the first and second divisions within 2 and 4 days, respectively. Furthermore, control (without cold pre-treatment) and maximum (12 days duration) low temperature pre-treatments showed lower protoplast yield and viability with no second divisions. The lowest protoplasm division frequency (1.4 ± 0.1%) was observed withoug cold pre-treatment.
Pollen Tetrad Protoplast Culture
After one day in culture, most of the regenerated protoplasts formed a smooth wall showing an elongated ovoid shape with the initiation of the first division occurring after 3 days of culture (Fig. 1e-f). Most of the protoplasts finished the first division after 7 days of culture. A few cells completed a second division resulting in 3–4 daughter cells with active cytoplasmic streaming. After 20 days of culture, multicellular structures were observed (Fig. 1g-h).
The Effect of Plant Growth Regulator Combinations on Cell Division Frequency
In five (S2, S3, S6, S7 and S8) of eight culture media tested, protoplasts regenerated a cell wall and cell division could be observed. The division frequency was highest and lowest in S3 and S6 (21.6 ± 2.4 and 3.4 ± 0.5% respectively) (Table 5). S3 promoted the shortest time of the first division time (2 days) followed by S8 (3 days); however, in this last no second division was observed. The presence of budding and brown cells showed the following pattern: S7 > S1-S2-S6-S8 > S3-S5 (Table 5). In S1, S4 and S5 medium no cell division took place. Therefore, according to the culture result, the favorable plant growth regulator combinations culture medium following pattern: S3 > S7 > S2 > S8 > S6 > S1-S4-S5.
Discussion
Haploids and doubled haploids are critical components of plant breeding (Dong et al. 2016). Haploid pollen tetrad protoplasts can be used as a starting material for the production of haploid plants (Bhojwani and Cocking 1972). They are also suitable for somatic genetics studies, since mutants would easily be detectable in haploids (Deka et al. 1977). In the present study, isolation of pollen tetrad protoplasts was achieved at >70% insolation rate. To our knowledge this is the first study in which a methodology to obtain sustained cell division from tetrad protoplasts has been reported.
Protoplast yield and division ability are affected by genotype and physiological status of the donor plants (Foulger and Jones 1986; Jones et al. 1989). Previous study has demonstrated that within an anther of Nicotiana tabacum cv. White Burley and N. sylvestris there is a gradient of tetrad development: older was at the base (notched end) and the younger towards the tip (Bhojwani and Cocking 1972). Compared to other studies which report a rate of nearly 100% protoplast isolation rate (Bajaj 1974; Bhojwani and Cocking 1972), the lower rate (74.6%) in this study may have been caused by the gradient of tetrad development within potato anthers. Because of the different developmental stages of tetrads, presumably older tetrads may have had spore walls more resistant to enzymatic degradation.
The snailase concentration and an appropriate osmoticum in the enzyme solution were crucial for optimal pollen tetrad protoplast isolation. Callose is an unbranched β-1,3 glucan and the snailase enzyme is richer in β-1,3 glucanase activity, and it is capable of digesting both the callose and to some extent cellulosic material (Bajaj 1974). When pollen tetrads were treated with enzyme solution mainly containing snailase, the callose around the tetrad began to dissolve within 15–20 min. The naked spores became spherical soon after their release from the tetrads, but those with cellulose already deposited on the wall took longer; the wall was gradually digested by the enzyme mixture after 2–4 h, resulting in a high yield of pollen tetrad protoplasts (Fig. 1c). The concentration of snailase is vital to the total protoplast yield and their cultivability, as judged by subsequent division. However, more than 1.2% of snailase concentration can damage tetrad protoplasts, resulting in lower viability, higher budding and multinuclei frequency during culture. In this study, snailase at 1.2% was most effective in release of intact protoplasts in all the four experimental systems examined (Table 3). In addition, the presence of an osmotic stabilizer was essential for adequate isolation frequency and viability of tetrad protoplasts. The use of enzyme solutions containing sucrose as osmoticum was better than mannitol, sorbitol or glucose in the enzyme solution for protoplast production leading to the highest division frequency and consecutive division ability (Table 3). Sucrose not only plays an important role in the balance and adjustment of medium osmoticum, but also providing the energy source for cell metabolism during cell culture (Yaseen et al. 2013).
Cold pre-treatment was vital for pollen tetrad protoplast isolation, as well as for cell division induction. Previous studies have reported that cell division induction frequency was driven by low temperature pre-treatments (4–5 °C) (Dong et al. 2016). Corroborating this report, we found that pre-treating the buds at 5 °C for 7d increased protoplast viability, division frequency (>20%) and cell division. This finding was similar to previous studies in the culture of Hemerocallis fulva uninucleate pollen protoplasts (Zhou 1989b). Cold pre-treatments to develop microspores have been used in a variety of plant species to promote genetic program switching from the development of gametophyte to sporophyte (He et al. 2006; Dong et al. 2016). The mechanisms of low temperature might also be appropriate for tetrad microspore protoplasts to divert the reproductive growth toward the vegetative, benefiting dedifferentiation during the subsequent culture.
The culture medium was a crucial factor to control the tetrad protoplast wall regeneration and its division initiation. In agreement with prior studies on pollen protoplast isolation and culture (Zhou 1989b; Xia et al. 1996; Li et al. 1992), we achieved a substantial improvement using the macro- and micronutrients from K3 medium, which included higher concentrations of Ca2+ and H2PO4 − presumably to stabilize cell membranes and maintain pollen protoplasts intact during the process. In addition, some amino acid supplements in culture medium can induce cell division and dedifferentiation (Nitsch 1974; Dong et al. 2016). In the present study, pollen tetrad protoplast regeneration, division and consecutive division were induced when culture medium was supplemented with glutamine and serine. Moreover, the combination of plant growth regulators was critical to promote cell division. The highest division and the best ability of successive division could be obtained in culture medium supplemented with 2,4-D 1.0 mg/L + KT 0.5 mg/L + 6-BA 0.4 mg/L. Therefore, the NAA in K3 basal medium was eliminated, observing a decrease in the number of budding cells during culture.
Previous research reported in vitro culture of pollen protoplasts resulted in cell wall regeneration, formation of tube-like and other abnormal structures and limited nuclear divisions (Bajaj 1974; Tanaka et al. 1987; Zhou 1988; Xia et al. 1996), however, embryogenic divisions, proembryo formation and uninucleate microspore clusters have been reported only in Hemerocallis fulva (Zhou 1989b). Although pollen protoplast induction to dedifferentiation and redifferentiation is an important problem that still needs to be solved, gamete somatic protoplast fusion has afforded a new way for rescuing or even ‘diverting’ pollen cells to the sporophytic pathway leading to producing hybrid or cybrid plants (Pirrie and Power 1986; Lee and Power 1988; Desprez et al. 1995). Present study revealed preliminary tetrad protoplasts division and multicellular colonies formation in vitro. The first division had equal and unequal division, both could continue to develop leading to organized or unorganized growth and a consecutive emergence of a diversity of morphologies and structures. Although the division of tetrad protoplasts were similar to the process of zygotic embrogenesis (Zhou 1989a), the regenerated plant was not obtained from the multicellular colonies. External factors, such carbohydrate or nitrogen starvation, high temperature and low pH can change gametophytic development of microspores to a sporophytic pathway (Custers et al. 1994; Touraev et al. 2001; Barinova et al. 2004). These results indicated that cold pre-treatment, plant growth regulators and amino acids (glutamine and serine) maybe the factors to induced the sporophytic developmental pathway in potato tetrad microspores.
Conclusions
In summary, an efficient and reproducible enzymatic maceration protocol was established to isolate living pollen tetrad protoplasts at high rates from Solanum tuberosum. This approach provides a baseline for further studies that may include gene transformation, fusion with somatic protoplasts, generation of haploid potato plants, offering a means to simplify the introgression of traits from new sources of genetic diversity. Because the culture of pollen tetrad protoplasts is complex and our protocol has resulted only in multicellular colonies to date, additional studies will be necessary to obtain regenerated plants from pollen tetrad protoplasts.
References
Arnalte, E., P. Pérez-Bermúdez, M.J. Cormejo, and J. Segura. 1991. Influence of microspore development on pollen protoplasts isolation in Digitalis obscura. Journal of Plant Physiology 138: 622–624.
Bajaj, Y.P.S. 1974. Isolation and culture studies on pollen tetrad and pollen mother-cell protoplast. Plant Science Letters 3: 93–99.
Bajaj, Y.P.S., and M.R. Davey. 1974. The isolation and ultrastructure of pollen protoplasts. In Fertilization in higher plants, ed. H.F. Linskens, 73–80. Amsterdam: Elsevier.
Barinova, I., C. Clément, L. Martiny, F. Baillieul, H. Soukupova, E. Heberle-Bors, and A. Touraev. 2004. Regulation of developmental pathways in cultured microspores of tobacco and snapdragon by medium pH. Planta 219: 141–146.
Barrell, P.J., S. Meiyalaghan, J.M.E. Jacobs, and A.J. Conner. 2013. Applications of biotechnology and genomics in potato improvement. Plant Biotechnology Journal 11: 907–920.
Bhojwani, S.S., and E.C. Cocking. 1972. Isolation of pollen protoplasts from pollen tetrads. Nature New Biology 239: 29–30.
Burlingame, B., B. Mouillé, and R. Charrondière. 2009. Nutrients, bioactive non-nutrients and anti-nutrients in potatoes. Journal of Food Composition and Analysis 22: 494–502.
Conner, A.J., J.M.E. Jacobs, and R.A. Genet. 1997. Transgenic potatoes versus ‘traditional’ potatoes: what's the difference? In McLean GD, ed. P.M. Waterhouse, G. Evans, and M.J. Gibbs, 23–36. Canberra: Cooperative research center for plant science and bureau of resource sciences.
Custers, J.B.M., J.H.G. Gordewener, Y. Nöllen, H.J. Dons, and M.M. Van Lookeren Campagne. 1994. Temperature controls both gametophytic and sporophytic development in microspore cultures of Brassica napus. Plant Cell Reports 13: 267–271.
Dai, C.X., D. Mertz, and V. Lambeth. 1987. Improved procedures for the isolation and culture of potato protoplasts. Plant Science 50: 79–84.
Deka, P.C., A.K. Mehra, N.N. Pathak, and S.K. Sen. 1977. Isolation and fusion studies on protoplasts from pollen tetrads. Experientia 33: 182–184.
Desprez, B., Y. Chupeau, and J.P. Bourgin. 1995. Preparation and fusion properties of protoplasts from mature pollen of Nicotiana tabacum. Plant Cell Reports 14: 199–203.
Dong, Y.Q., W.X. Zhao, X.H. Li, X.C. Liu, N.N. Gao, J.H. Huang, W.Y. Wang, X.L. Xu, and Z.H. Tang. 2016. Androgenesis, gynogenesis, and parthenogenesis haploids in cucurbit species. Plant Cell Reports. 35: 1991–2019.
Fan, L.M., Y.F. Wang, H. Wang, and W.H. Wu. 2001. In vitro Arabidopsis pollen germination and characterization of the inward potassium currents in Arabidopsis pollen grain protoplasts. Journal of Experimental Botany 52: 1603–1614.
Fang, K., L. Zhang, and J. Lin. 2006. A rapid, efficient method for the mass production of pollen protoplasts from Pinus bungeana Zucc. Ex Endl. And Picea wilsonii mast. Flora 201: 74–80.
Ferrie, A.M.R., and K.L. Caswell. 2011. Isolated microspore culture techniques and recent progress for haploid and doubled haploid plant production. Plant Cell, Tissue and Organ Culture 104: 301–309.
Foulger, D., and M.G.K. Jones. 1986. Improved efficiency of genotype-dependent regeneration from protoplasts of important potato cultivars. Plant Cell Reports 5: 72–76.
Germanà, M.A. 2011. Anther culture for haploid and doubled haploid production. Plant Cell, Tissue and Organ Culture 104: 283–300.
Germanà, M.A., and B. Chiancone. 2001. Gynogenetic haploids of citrus after in vitro pollination with triploid pollen grains. Plant Cell, Tissue and Organ Culture 66: 59–66.
He, G., J. Zhang, K. Li, Z. Xiong, M. Chen, J. Chang, Y. Wang, G. Yang, and B. Barnabás. 2006. An improved system to establish highly embryogenic haploid cell and protoplast cultures from pollen calluses of maize (Zea mays L.). Plant Cell. Tissue and Organ Culture 86: 15–25.
Hijmans, R.J. 2001. Global distribution of the potato crop. American Journal of Potato Research 78: 403–412.
Jones, H., A. Karp, and M.G.K. Jones. 1989. Isolation, culture, and regeneration of plants from potato protoplasts. Plant Cell Reports 8: 307–311.
Kao, K.N., F. Constabel, M.R. Michayluk, and O.L. Gamborg. 1974. Plant protoplasts fusion and growth of intergeneric hybrid cells. Planta 120: 215–227.
Lee, C.H., and J.B. Power. 1988. Intraspecific gametosomatic hybridization in Petunia hybrida. Plant Cell Reports 7: 17–18.
Li, S.Q., H.Y. Yang, and C. Zhou. 1992. Release of pollen protoplasts in large quantities in Brassica napus and B. campestris Var. purpurea. Acta Botanica Sinica 34: 339–345.
Loewus, F.A., B.G. Baldi, V.R. Franceschi, L.D. Meinert, and J.J. McCollum. 1985. Pollen sporoplasts: dissolution of pollen walls. Plant Physiology 78: 652–654.
Muthoni, J., J. Kabira, H. Shimelis, and R. Melis. 2015. Tetrasomic inheritance in cultivated potato and implications in conventional breeding. Australian Journal of Crop Science 9: 185–190.
Nitsch, C. 1974. La culture de pollen isolé sur milieu synthétìque. Comptes Rendus de l'Académie des Sciences 278 (D): 1031–1034.
Ortiz, R. 1998. Potato breeding via ploidy manipulations. Plant Breeding Reviews 16: 15–86.
Pirrie, A., and J.B. Power. 1986. The production of fertile, triploid somatic hybrid plants (Nicotiana glutinasa (n) + N.tabacum (2n)) via gametic: somatic protoplast fusion. Theoretical and Applied Genetics 72: 48–52.
Rajasekhar, E.W. 1973. Nuclear divisions in protoplasts isolated from pollen tetrads of Datura metel. Nature 246: 223–224.
Tanaka, I., C. Kitazume, and M. Ito. 1987. The isolation and culture of lily pollen protoplasts. Plant Science 50: 205–211.
Tavazza, R., and G. Ancora. 1986. Plant regeneration from mesophyll protoplasts in commercial potato cultivars (Primura, Kennebec, Spunta, Desirée). Plant Cell Reports 5: 243–246.
Touraev, A., M. Prosser, and E. Heberle-Bers. 2001. The microspore: a haploid multipurpose cell. Advances in Botanical Research 35: 53–109.
Wenzel, G., O. Schieder, T. Przewozny, S.K. Sopory, and G. Melchers. 1979. Comparison of single cell culture derived Solanum tuberosum L. plants and a model for their application in breeding programs. Theoretical and Applied Genetics 55: 49–55.
Xia, H.J., C. Zhou, and H.Y. Yang. 1996. Isolation of young pollen protoplasts in Nicotiana tobacum and its early developmental pathway in vitro. Acta Botanica Sinica 38: 113–117.
Yaseen, M., T. Ahmad, G. Sablok, A. Standardi, and A.I. Hafiz. 2013. Review: role of carbon sources for in vitro plant growth and development. Molecular Biology Reports 40: 2837–2849.
Yoo, S.D., Y.H. Cho, and J. Sheen. 2007. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nature Protocols 2: 1565–1572.
Zhang, F., D. Wang, and Y.P. Wang. 2004. Isolation of pollen protoplasts in Solanum tuberosum. Scientia Agricultura Sinica 37: 1358–1362.
Zhou, C. 1988. Mass isolation and culture of pollen protoplasts in three plant species. Acta Botanica Sinic 30: 362–367.
Zhou, C. 1989a. A study on isolation and culture of pollen protoplasts. Plant Science 59: 101–108.
Zhou, C. 1989b. Cell divisions in pollen protoplasts culture of Hemerocallis fulva L. Plant Science 62: 229–235.
Acknowledgments
This work was supported by Program for National Natural Science Foundation (31171477, 31471433; 31060063; 31260094), Gansu National Science Foundation (1506RJZA013), Gansu High Educational Scientific Special Project and Gansu Provincial Youth Science and Technology Foundation Project (1606RJYA229). Authors thank Dr. David Ramirez to revise and edit the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, Y., Cheng, L., Liang, Y. et al. Isolation and Culture of Pollen Tetrad Protoplasts from Solanum tuberosum . Am. J. Potato Res. 94, 417–424 (2017). https://doi.org/10.1007/s12230-017-9578-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12230-017-9578-0