
Abstract
Next-generation sequencing methods provide comprehensive data for the analysis of structural and functional analysis of the genome. The draft genomes with low contig number and high N50 value can give insight into the structure of the genome as well as provide information on the annotation of the genome. In this study, we designed a pipeline that can be used to assemble prokaryotic draft genomes with low number of contigs and high N50 value. We aimed to use combination of two de novo assembly tools (SPAdes and IDBA-Hybrid) and evaluate the impact of this approach on the quality metrics of the assemblies. The followed pipeline was tested with the raw sequence data with short reads (< 300) for a total of 10 species from four different genera. To obtain the final draft genomes, we firstly assembled the sequences using SPAdes to find closely related organism using the extracted 16 s rRNA from it. IDBA-Hybrid assembler was used to obtain the second assembly data using the closely related organism genome. SPAdes assembler tool was implemented using the second assembly, produced by IDBA-hybrid as a hint. The results were evaluated using QUAST and BUSCO. The pipeline was successful for the reduction of the contig numbers and increasing the N50 statistical values in the draft genome assemblies while preserving the coverage of the draft genomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fast development of high-throughput sequencing technologies has advanced the rate and quality of sequencing, particularly for prokaryotes. Short or long reads produced by next-generation sequencing can provide comprehensive results using accurate bioinformatics approaches. If one organism has got a reference genome in the database, genome data is mapped against reference genome. If there is, however, no reference genome in the database, de novo assembly approach is performed using various assembler tools. However, there are still many challenges to obtain a complete genome and/or a nearly finished draft genome. Challenges for obtaining high-quality data raise from limitations of computational tools errors in sequence reads and genomic background of the organisms (e.g., distribution of repeating regions, GC content) (Page et al. 2016).
Obtaining sufficient data to obtain complete genome sequence requires using more than one platform for sequencing which in turn increases the cost. Although new technologies (e.g., Nanopore) offer high-quality long reads, there are thousands of draft genomes with a high number of contigs deposited in the databases that can provide extensive amount of data when their quality would be increased (Earl et al. 2011).
Short or long reads are processed through the assembly tools to produce genomic data. Choosing either short or long reads for genome assembly comes with different pitfalls. While short reads provide more accurate data, error rate is higher for long reads, especially at the homopolymer regions (Utturkar et al. 2017; Liao et al. 2019). On the other hand, while short reads are inadequate for assembling the repetitive regions, long reads can provide more reliable data (Page et al. 2016). To overcome these drawbacks, using both the short and the long reads is processed together under polishing methods to obtain the complete genome or proofreading using short reads for long reads beforehand (Utturkar et al. 2017). Even though polishing method provides accurate results, it is not encouraged due to the high cost. Mate pair sequencing is an alternative approach where long-insert paired-end DNA libraries are used to obtain the complete genome.
Many studies are contented with draft genome data for especially prokaryotes since the draft genomes can provide adequate numbers of contigs to perform annotation analysis of highly conserved sequences (Ricker et al. 2012). Increasing the quality and quantity of the contigs representing the draft genome will fortify the data for downstream analyses for comparative genomics approaches; in addition, it provides more comprehensive functional annotation results. It may be challenging to distinguish evolutionary close bacterial species due to high similarity of 16 s rDNA region. Thus, in addition to 16 s rDNA region, average nucleotide identity (ANI) scores have been evaluated simultaneously to increase the resolution power for delineating the exact species names (Kim et al. 2014). ANI scores are simply the estimates of the average nucleotide identity between two genomic datasets (Goris et al. 2007).
In addition, draft genomes can also assist the construction of whole genome sequence maps. Reference-assisted/reference-guided assembly pipelines have been studied to obtain complete genomes (Kolmogorov et al. 2014; Guizelini et al. 2016). To provide whole genome sequence, the assembly is ordered against reference genome, and gaps are closed by alternative assemblies that have been produced using other assemblies (Guizelini et al. 2016). On the other hand, the reference-assisted assembly approaches have some biases since the reference genomes may have some errors and genome rearrangement among the species (Lischer and Shimizu 2017). The success of this approach is limited by the quality of the raw data and/or reference genome. Therefore, construction of high-quality draft genomes is crucial not only for annotation studies but also for obtaining complete genomic sequences.
According to NCBI prokaryotes genome, a total of 21,857 complete genomes and 275,100 draft bacterial genomes are listed as of February 10, 2021. The listed draft genomes are represented by 167,258 contig-level and 107,842 scaffold-level data. Detailed evaluation of the available draft genomes show that almost half of the data are represented by higher than 100 contigs (National Center for Biotechnology Information (NCBI) 1988). Even if the draft genomes are enough to carry out downstream analysis, it is, nevertheless, important to obtain less fragmented genome as well. These statistics in NCBI database show that the high number of contigs in the draft genomes can be addressed in terms of lack of accurate bioinformatic approaches.
In this study, we focused on improving the outcomes of de novo assembly by obtaining high-quality draft genomes with lower contig number. We designed a pipeline by combining two powerful assembly tools. To prove the strength of the approach, we run the pipeline using the raw data for ten previously assembled draft genomes and compared the quality matrices between the studies.
Methods
Data preprocessing
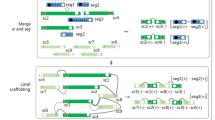
In this study, short pair-end Illumina data were used from four independent studies that were previously assembled at draft genome level. We retrieved the raw Illumina short pair-end read data and the assembled draft genomes from NCBI database repositories for 5 g-negative and 5 g-positive bacteria with a ranging GC content from 30.5 to 66.4. The accession numbers of raw data and original genome assemblies for the datasets are provided in Table 1. Metadata for draft genomes of the species included in the study is provided in Online Resource 1. The scripts that can be adapted to follow the pipeline are provided in Online Resource 2. The summary of the preprocessing steps in the pipeline is shown in Fig. 1a. Quality check for the raw data was carried out using FASTQC (v0.11.3) tool (Andrews 2010). For trimming and quality filtering, we used Trimmomatic tool (v0.39) (Bolger et al. 2014). Depending on the quality metrics of the data, the parameters were adjusted to remove the adaptors and low-quality reads from the 5′ and/or 3′ ends of the reads: LEADING:3, TRAILING:3, SLIDINGWINDOW:4:15|20 MINLEN:50.

Workflow of combined and reference-assisted de novo assembly approach. The pipeline summarizes the steps from preprocessing (a) to obtaining the first assembly (b), draft assembly (c), and final assembly (d), respectively
SPAdes v. 3.14.1 (Bankevich et al. 2012) was used to obtain the first assembly. The assembly data obtained from each dataset was used to predict the most related species bacterial species. 16 s rRNA region was parsed from the assembly using barrnap (v0.9) tool (Seemann 2013). BLAST analysis was used for the prediction of bacterial species showing the highest similarity to the parsed query sequences against “Refseq Representative genomes (refseq_representative_genomes)” and/or “Refseq Genome Database (refseq_genomes).”
We downloaded the genome data after evaluating the similarities of 16S rRNA sequences and the availability of the corresponding genome data. These genomes were then used as a reference genome in the downstream assembly steps (Fig. 1b) to produce the draft assembly.
De novo assembly
IDBA-Hybrid, as the extension of IDBA-UD, is an assembler tool that works based on de Bruijn algorithm and improves the assembly using a guide reference genome (Peng et al. 2012). To perform the alignment and find similar regions, the similarity parameter was adjusted to ≥ 0.95 for aligning the sequences and finding the similar regions. For each dataset, the predicted reference genome was selected for IDBA-Hybrid reference parameter. The preprocessed datasets were selected to obtain the draft genome by using IDBA-Hybrid v1.1.3 with the default parameters. In this study, the draft assemblies produced by IDBA-Hybrid (Fig. 1c) were used for each corresponding dataset.
Final assembly was carried out using SPAdes v. 3.14.1. Filtered paired-end reads were assembled adjusting the parameters as k-mer: 21, 33, 55, 77, 99, and 127; trusted-contigs: true; only-assembler: true; and cov-cutoff: auto. “Trusted contigs” is the parameter to use an assembly that was previously assembled in a different assembly tool to provide a guidance during de novo assembly. However, it is the parameter for guiding the assembly data input, not for mapping to the genome per se (Prjibelski et al. 2020). Only-assembler parameter was selected to run the assembly module only since the data was quality-filtered in previous steps (Fig. 1d).
Evaluation of final assemblies
To evaluate the quality of final assemblies obtained in this study, we downloaded the original assembly files deposited in NCBI GenBank database. Quality metrics (number of contigs, size of largest contig, total length of draft genome, GC content, N50 value, and L75 value) for the final assemblies were compared against originally assembled corresponding genomes using QUAST (v5.0.2) (Gurevich et al. 2013). To assess the completeness of the final genomes, single-copy orthologs in the final assemblies were compared to the genomes against the original genomes using BUSCO (v4.1.4) (Fig. 1d) (Simão et al. 2015). In addition, we assessed the correctness of final assemblies using Reapr (v1.0.18) (Hunt et al. 2013). We used the following formula to calculate REAPR scores (Earl et al. 2011).
Results and discussion
In this study, we introduced a pipeline to improve the outcomes of de novo assembly in the bacterial genomes. De novo assembly is crucial for obtaining genome data for downstream bioinformatics analysis while working with the organisms without any annotated reference genome. It is usually challenging to obtain the complete genome data using short reads through one sequence platform. Thus, obtaining a good quality draft genome will be satisfying by benefiting both cost and time. To improve the quality of draft genomes, many genomes have been deposited in the public databases, and they can allow us to perform reference-assisted and combined tool approach. In this study, we used two different assembler tools, IDBA-Hybrid with reference parameter and SPAdes with trusted-contigs parameter since SPAdes requires only contig-level input as a hint to produce genomic data. We aimed to use IDBA-Hybrid with a closely related genome to produce contigs file for the input of trusted-contigs parameter in SPAdes. In other words, we prepared a hint assembly data using IDBA-Hybrid to run it with SPAdes. This hint file produced from IDBA-Hybrid was used for graph construction, gap closure, and repeat resolution when SPAdes run with trusted-contigs parameter. This implies that in the final step to produce the final draft genomes, we did not map reads against the second assembly produced from IDBA-Hybrid to do de novo assembly. Thus, we obtained a more compact draft genome using short reads than already published in the database. We tested the pipeline on the data deposited in the NCBI database belonging four independent studies covering six species classified under four different genera: Pseudomonas, Bacillus, Burkholderia, and Clostridium.
In prokaryotes, 16 s rRNA region is used for phylogenetic analysis since it is conserved among the species. After the quality filtering of the raw data, we obtained the first assembly by standard de novo assembly approach using SPADES tool. The first assembly data was used to parse the 16 s rRNA region before performing the reference-assisted de novo assembly to obtain the draft assembly by comparing 16 s rRNA regions. Since no reference genomes are available for the species of interest, we predicted the bacterial species with high similarity and available complete genome by BLAST analysis (Table 2). We choose complete genomes with the best hit as reference genomes through visual assessment in both “RefSeq Representative” and “RefSeq Genomes” databases (Online Resource 3). Since the pipeline uses the reference genome as a hint, assembly is not expected to be dramatically affected if selected reference genome similarity is high, although it does not have the best hit.
In addition, prediction of average nucleotide identity (ANI) scores is also effective in estimating the similarity of certain bacteria, among others. However, considering that there could be too many genomes, it may take more time to calculate ANI scores. If it is designed an optimized process of downloading the genomes and calculating ANI scores, it would obviously give more robust result to determine the closely related organism.
We obtained the draft assemblies using corresponding genome data for each dataset. The purpose of the draft assembly is to act as a guiding reference for de novo assembly. We analyzed the quality of the assembled draft genomes comparing to originally published data and the early assemblies (Fig. 1b and c) obtained using the pipeline. Quast analysis was used to assess the contig metrics along with GC content and N50 and L75 statistical values (Table 3). QUAST report assessment showed that using the proposed pipeline significantly improved the quality metrics of draft genome assemblies. We were able to decrease the number of the contigs down to 50.4% (Table 3). The highest reduction in the contigs was obtained for Po_2 (new) by a reduction of 62 contigs, while the lowest reduction was obtained for Bs_2 (new) that only one contig less draft genome was assembled. On the other hand, we presented statistics of mapping rate of reads onto the final assemblies. This statistic showed that reads aligned onto the final assemblies over 99%, except to one is 98.13% (Table 4). All assemblies after completing the pipeline provided better quality compared to the assemblies obtained from IDBA-Hybrid. When compared to the first assemblies from SPAdes (Fig. 1b), most of the final assemblies showed better quality. Interestingly, while the first assembly of Ps_2 provided slightly higher N50 value and contig number, Po_1 provided higher N50 value and slightly less contig number. Full QUAST report has been presented in the Online Resource 4.
On the other hand, N50 statistics showed significant increment among the assemblies (Table 3). The highest increment in N50, the same species, Po_2 (new), increased from 121,728 to 410,410 kb as 70.3%, while the lowest increment was occurred in Bs_3 that was from 22,386 to 22,678 as 1.3%. The most efficient reduction in the contigs and the increment in N50 have occurred in Pseudomonas species. GC contents of the species changed only at the decimal level. In addition to the number of contigs and N50, the largest contiguous length in the assemblies increased except one species—Bs_2 (new) (Table 3, Online Resource 4). Our approach was also effective in Bacillus sporothermodurans strains. Bacillus strains are known with high number of repeating elements (Kunst et al. 1997; Økstad et al. 2004). Aligning of short reads with repeating regions is challenging especially for the read lengths shorter than that of the repeating regions. Even though the effectiveness is less compared to the results that we obtained for other species, we were still able to obtain improved draft genome for Bacillus sp.
Since our results provided draft genomes with less contig number, we evaluated the completeness of the final genomes to assess if there is any negative impact on the coverage of the contigs. We evaluated the BUSCO metrics using a total of seven databases covering kingdom, phylum, and order level information. We selected the Firmicutes phylum for Bacillus and Clostridium, the Proteobacteria phylum for Pseudomonas and Burkholderia, Bacillales order for Bacillus, Clostridia order for Clostridium, Pseudomonadales order for Pseudomonas, and Burkholderiales order for Burkholderia. Our BUSCO results showed that the genomes we produced have less fragmented and/or same completeness with the original genomes. As the completeness is evaluation of the assembly, the higher completeness level can give better annotation results in the downstream analysis.
In BUSCO analysis, bacteria kingdom dataset show that some of the fresh assemblies were more complete and/or less fragmented in Fig. 2. In Firmicutes, Bacillales, and Clostridiales, datasets show that there were no notable changes among both assemblies in Fig. 3. Similar results are observed in Proteobacteria, Pseudomonadales, and Burkholderiales datasets in Fig. 4. It shows that the proposed pipeline did not reduce any quality of the coverage while reducing the number of the contigs. According to the correctness analysis, small differences were observed among all the assemblies. All REAPR statistics have been presented in Online Resource 5. Thus, the pipeline does not improve nor reduce the correctness of the assemblies while reducing the contig numbers.

BUSCO assessments and comparison of the genome assemblies’ completeness in a set of the 124 genes in bacteria kingdom

BUSCO assessments and comparison of completeness for the genome assemblies. Bacillus and Clostridium species have been analyzed in a set of 218 genes in Firmicutes database (a); Bacillus species have been analyzed in a set of 450 genes in Bacillales database (b). Clostridium species have been analyzed in a set of 264 genes in Clostridiales database (c)

BUSCO assessments and comparison of completeness for the genome assemblies. Pseudomonas and Burkholderia species have been analyzed in a set of 219 genes in Proteobacteria database (a); Pseudomonas species have been analyzed in a set of 782 genes in Pseudomonadales database (b); Burkholderia species have been analyzed in a set of 688 genes in Burkholderiales database (c)
According to REAPR summary score (Fig. 5, Online Resource 5) and N50 and contigs number in the QUAST report (Table 3, Online Resource 4), Burkholderia and all Bacillus sp. were always improved by our proposed pipeline. As aforementioned, the first assemblies of Ps_1 and Po_1 were slightly better than final assemblies. While Ce (new), Ps_1 (new) and Po_2 (new) had better N50 and contigs number, the first assemblies of them had higher REAPR summary score. Since Burkholderia lata has 3 chromosomes (Bugrysheva et al. 2016; Leong et al. 2018), and Bacillus sp. involves repetitive regions in complex genome structures (Kunst et al. 1997; Økstad et al. 2004), it is likely that the pipeline is particularly more effective on more repetitive and complex genomes. Although there are many concerns about the draft genome that is produced through reference-assisted technique due to genome rearrangement and possibility errors in reference genome, we did not perform gene ordering or change the structure of the genome. We used IDBA-Hybrid, which finds similar regions from the closely related genome with a 95% confidence level, to help to do de novo assembly using the reads to produce the second assembly. Those second assemblies were used to provide the hints to run SPAdes. This implies that we used similar assemblies produced by different assembler software, IDBA-Hybrid, when we run SPAdes. There are different tools and pipelines offered to overcome the challenges for de novo assembly of short reads. Researchers often choose de novo assembly due to lack of reference genomes. In addition, the reference-assisted assembly approaches may show biases because of the errors in the reference genomes (Earl et al. 2011; Lischer and Shimizu 2017). It was already suggested that de novo assemblies can benefit from the combination with reference mapping. In the previous studies, promising results were obtained by combination of de novo assembly with reference mapping in eukaryotic genomes (Bradnam et al. 2013; Lischer and Shimizu 2017). In another study, scientists effectively improved the quality of prokaryotic draft assemblies by post-processing the assemblies through Ragout pipeline (Kolmogorov et al. 2014). It is suggested that IDBA was the most outperforming assembly tool for small genomes with uneven coverage (Peng et al. 2012). In previous study, it was one of the most outperforming tools as a reference-guided de novo assembly approach where IDBA was used for de novo assembly after obtaining superblocks via Bowtie2 (Lischer and Shimizu 2017). In our study, we contribute the field by proposing a pipeline to improve the quality of prokaryotic draft genomes starting from short raw sequences by integrating SPAdes and IDBA-Hybrid.

REAPR summary scores for the genome assemblies. The score was calculated by multiplying the number of error free bases with square of N50 contig broken length divided by N50 contig length
Conclusion
De novo assembly is not only complex and challenging but also a crucial step before downstream analysis of the organisms. In this study, we presented a pipeline for combined tools and reference-assisted approaches that improve de novo assembly and construct a high-quality draft genome in prokaryotes. Our approach can be used as a promising option for performing improved de novo assembly in prokaryotes. Developing the automated pipeline is difficult due to the challenges in data preprocessing and k-mers optimization and choosing closely related organism’s genome, but implementing the proposed pipeline to an automated version would provide advantages for the future studies to present user-friendly pipeline. In this study, we provided the analysis results for bacteria classified under different genera. Indeed, the pipeline can benefit from the analysis of more genomes when the automated version is implemented and run with large datasets on high-performance computing clusters.
Third-generation sequencing technologies are most likely to be dominant in the future as the accuracy rate increases for homopolymer regions. However, the approach we presented can be considered as an alternative for the low depth or complex genomes that have been encountered from short reads through one platform. In the future, the pipeline can be improved by including additional tools for assembly of eukaryotic genomes as these genomes generally are more difficult to assemble due to complexity and/or heterozygosity.
References
Andrews S (2010) FASTQC A quality control tool for high throughput sequence data. In: Babraham Inst. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Bankevich A, Nurk S, Antipov D et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. https://doi.org/10.1089/cmb.2012.0021
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. https://doi.org/10.1093/bioinformatics/btu170
Bradnam KR, Fass JN, Alexandrov A et al (2013) Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience. https://doi.org/10.1186/2047-217X-2-10
Bugrysheva JV, Cherney B, Sue D et al (2016) Complete genome sequences for three chromosomes of the Burkholderia stabilis type strain (ATCC BAA-67). Genome Announc. https://doi.org/10.1128/genomeA.01294-16
Earl D, Bradnam K, St. John J et al (2011) Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res 21
Esmaeel Q, Issa A, Sanchez L et al (2018) Draft genome sequence of Burkholderia reimsis BE51, a plant-associated bacterium isolated from agricultural rhizosphere. Microbiol Resour Announc. https://doi.org/10.1128/mra.00978-18
Goris J, Konstantinidis KT, Klappenbach JA et al (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. https://doi.org/10.1099/ijs.0.64483-0
Guizelini D, Raittz RT, Cruz LM et al (2016) GFinisher: a new strategy to refine and finish bacterial genome assemblies. Sci Rep. https://doi.org/10.1038/srep34963
Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics. https://doi.org/10.1093/bioinformatics/btt086
Hollmann J, Brinks E, Schwake-Anduschus C et al (2019) Draft genome sequences of Pseudomonas sp. strains isolated from wheat in Germany. Microbiol Resour Announc https://doi.org/10.1128/mra.00178-19
Hunt M, Kikuchi T, Sanders M et al (2013) REAPR: a universal tool for genome assembly evaluation. Genome Biol. https://doi.org/10.1186/gb-2013-14-5-r47
Kim M, Oh HS, Park SC, Chun J (2014) Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. https://doi.org/10.1099/ijs.0.059774-0
Kolmogorov M, Raney B, Paten B, Pham S (2014) Ragout - a reference-assisted assembly tool for bacterial genomes. Bioinformatics. https://doi.org/10.1093/bioinformatics/btu280
Kunst F, Ogasawara N, Moszer I et al (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390:249–256. https://doi.org/10.1038/36786
Leong LEX, Lagana D, Carter GP et al (2018) Burkholderia lata infections from intrinsically contaminated chlorhexidine Mouthwash, Australia, 2016. Emerg Infect Dis 24
Liao X, Li M, Zou Y et al (2019) Current challenges and solutions of de novo assembly. Quant Biol
Lischer HEL, Shimizu KK (2017) Reference-guided de novo assembly approach improves genome reconstruction for related species. BMC Bioinformatics. https://doi.org/10.1186/s12859-017-1911-6
National Center for Biotechnology Information (NCBI) (1988) Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. https://www.ncbi.nlm.nih.gov/genome. Accessed 2 Sep 2020
Økstad OA, Tourasse NJ, Stabell FB et al (2004) The bcr1 DNA repeat element is specific to the Bacillus cereus group and exhibits mobile element characteristics. J Bacteriol 186:7714–7725. https://doi.org/10.1128/JB.186.22.7714-7725.2004
Owusu-Darko R, Allam M, de Oliveira SD et al (2019) Genome sequences of Bacillus sporothermodurans strains isolated from ultra-high-temperature milk. Microbiol Resour Announc. https://doi.org/10.1128/mra.00145-19
Page AJ, De Silva N, Hunt M et al (2016) Robust high-throughput prokaryote de novo assembly and improvement pipeline for Illumina data. Microb Genomics. https://doi.org/10.1099/mgen.0.000083
Palevich N, Palevich FP, Maclean PH et al (2019) Draft genome sequence of Clostridium estertheticum subsp. laramiense DSM 14864T, isolated from spoiled uncooked beef. Microbiol Resour Announc. https://doi.org/10.1128/mra.01275-19
Peng Y, Leung HCM, Yiu SM, Chin FYL (2012) IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. https://doi.org/10.1093/bioinformatics/bts174
Prjibelski A, Antipov D, Meleshko D et al (2020) Using SPAdes de novo assembler. Curr Protoc Bioinforma. https://doi.org/10.1002/cpbi.102
Ramasamy KP, Telatin A, Mozzicafreddo M et al (2019) Draft genome sequence of a new Pseudomonas sp. Strain, ef1, associated with the psychrophilic antarctic ciliate Euplotes focardii. Microbiol Resour Announc. https://doi.org/10.1128/mra.00867-19
Ricker N, Qian H, Fulthorpe RR (2012) The limitations of draft assemblies for understanding prokaryotic adaptation and evolution. Genomics. https://doi.org/10.1016/j.ygeno.2012.06.009
Seemann T (2013) barrnap 0.9 : rapid ribosomal RNA prediction. Github.Com
Simão FA, Waterhouse RM, Ioannidis P et al (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. https://doi.org/10.1093/bioinformatics/btv351
Utturkar SM, Klingeman DM, Hurt RA, Brown SD (2017) A case study into microbial genome assembly gap sequences and finishing strategies. Front Microbiol. https://doi.org/10.3389/fmicb.2017.01272
Author information
Authors and Affiliations
Contributions
Uğur Çabuk contributed to study conception and design, data analysis, evaluation of results, and writing/editing the manuscript. Ercan Selçuk Ünlü contributed to study conception and design, mentoring Uğur Çabuk throughout the data analysis, evaluation of results, and writing/editing the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Çabuk, U., Ünlü, E.S. A combined de novo assembly approach increases the quality of prokaryotic draft genomes. Folia Microbiol 67, 801–810 (2022). https://doi.org/10.1007/s12223-022-00980-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12223-022-00980-7