
Abstract
Hemoglobinopathies are monogenic diseases with a significant impact on public health. It is estimated that 5.2% of the world population has at least one genetic alteration in the genes that code for globins and in Africa, 18.2% of the population have hemoglobinopathies of clinical relevance. Using hemoglobin capillary electrophoresis, we screen newborns from the Central Maputo Hospital in Mozambique for hemoglobinopathies and then used various PCR to genotype-affected newborns. Screening of newborns revealed that 35.8% had hemoglobinopathy traits, with variants running in the position of Hb Bart (33.2%), Hb C (1.7%), Hb D (0.8%), and Hb S (sickle cell) (0.4%). The molecular test identified that 9.9% of the newborns were homozygous for 3.7 kb and/or 4.2 kb deletion genotype and that there may be an uncharacterized deletion of MCS-R located on chromosome 16. This study also confirmed the presence of hemoglobin variants, Hb Maputo and Hb S, through the identification of their defining mutations. No mutations for Hb C were found; thus, we believe that it was a result of a screening error or contamination. This study successfully identified hemoglobinopathies of clinical significance within the Mozambican population, α-thalassemia and hemoglobin S, as well as their associated genotypes. Future research should include samples from other regions of the country and the use of a more comprehensive molecular diagnostic tool. With the confirmation of clinically significant genotypes, the local health system should promote the importance of screening of hemoglobinopathies in Mozambican newborns and adults.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Hemoglobinopathies are monogenic diseases with a significant impact on public health (Modell and Darlison 2008). These genetic diseases are originating in tropical and subtropical regions, but it has spread throughout the world (Modell and Darlison 2008). They are classified into two categories, namely variant hemoglobin and as thalassemia (Loukopoulos 2003; Modell and Darlison 2008). Variant hemoglobin (Hb) are a result of genetic alterations that change one of the amino acids in the α and/or the β subunit (Modell and Darlison 2008; Loukopoulos 2003). While thalassemia occurs due to insufficient (α + /β +) or the absence (α0/β0) synthases of the α and/or the β subunits (Loukopoulos 2003; Modell and Darlison 2008). From the clinical perspective, the relevant disorders are severe thalassemia, and the trait of sickle cell anemia (Hb S), Hb C, Hb D, and Hb E (Loukopoulos 2003; Modell and Darlison 2008; Goonasekera et al. 2018).
1.1 Hemoglobin: structure and gene expression
Adult hemoglobin (Hb A—α2β2 and Hb A2—α2δ2) is a tetramer composed of two α chains and two β chains, with a heme group at the center in each globin chain and is responsible for oxygen transportation within the human body. During the embryonic phase, three distinct hemoglobins are produced: Hb Gower 1 (ζ2γ2), Hb Gower 2 (α2ε2) and Hb Portland (ζ2γ2), that is replaced by fetal hemoglobin (Hb F—α2γ2) during the fetal. Fetal hemoglobin is the predominant hemoglobin at the time of birth and decreases over the first year (Loukopoulos 2003; Goonasekera et al. 2018; Mettananda and Higgs 2018).
The synthesis of hemoglobin is regulated by two clusters of genes, globin α genes and globin β genes located on chromosomes 16 and 11, respectively (Mettananda and Higgs 2018). The α genes are located on chromosome 16 (16p13.3) and contain the genes of the Hb subunit α1 (HBA1), Hb subunit α2 (HBA2), globin ζ2 and globin θ, along with three pseudogenes (HBPZ1/ѱζ, ψα1 e ψα2) (Galanello and Cao 2011; Farashi and Harteveld 2018). The genes are organized in order of gene expression and regulated by four multispecies control regions (MCS-R1-4). α thalassemia is usually the result of large deletions in the hemoglobin α region on chromosomes 16 (Farashi and Harteveld 2018; Mettananda and Higgs 2018). Deletions in this region (represented as “-”) cause changes in the synthesis of each α chain resulting in the minor (-α/αα) and major (-α/-α or -αT/αα) thalassemia, Hb H (--/-α) and Hb Bart (--/-- or αT-/--) genes (Galanello and Cao 2011; Farashi and Harteveld 2018). α-thalassemia can be caused by non-deletional α-gene mutations (αT), that frequently occur in the MCS-R1-R4 (Galanello and Cao 2011; Farashi and Harteveld 2018; Goonasekera et al. 2018; Mettananda and Higgs 2018).
The β globin genes are located on chromosome 11 (11p15.6), comprise of the β subunit Hb (HBB), ε globin, Gγ globin, Aγ globin, δ globin genes and a pseudogene (ѱβ1). This group is organized in the order of gene expression and regulated by five locus control regions (LCR). Changes in the HBB gene or the LCR can result in β-thalassemia (β0, β+, β++), and variant hemoglobin such as Hb S (β6 glutamic acid → valine), Hb C (β6 glutamic acid → lysine), Hb E (β26 glutamic acid → valine), Hb DPunjab (β121 glutamic acid → Glutamine), and Hb OArab (β121 glutamic acid → lysine). Unlike α-thalassemia, β-thalassemia and variant hemoglobin are caused by point mutations, and, currently, more than 250 of these mutations have been identified (Hardison et al. 1998; Loukopoulos 2003; Goonasekera et al. 2018; Mettananda and Higgs 2018).
In both cases, the genotype determines the clinical phenotype of hemoglobinopathies, whether an individual is a carrier or if they suffer from mild, moderate, or severe anemia (Goonasekera et al. 2018; Mettananda and Higgs 2018).
1.2 Diagnostics
The confirmatory diagnosis of the hemoglobinopathies is established using the following techniques: hemoglobin electrophoresis, capillary electrophoresis, isoelectric focusing (IEF), and high-performance liquid chromatography (HPLC), where HPLC and IEF are the most used (Sabath 2017; Goonasekera et al. 2018). Two disadvantages of these techniques are their low sensitivity in diagnosing β-thalassemia, especially in newborns, and the inability to identify carriers of hemoglobinopathies (Sabath 2017). The severity of the phenotype is determined by the genotype and due to scientific progress in the last 30 years in molecular biology, there are several techniques for the genotyping of hemoglobinopathies, such as sequencing, polymerase chain reaction (PCR), gap -PCR, and real-time PCR (Vrettou et al. 2003; Liu et al. 2006; Benson and Therrell 2010; Munkongdee et al. 2010; Sabath 2017). Although these techniques are not applied in routine for diagnosis, due to their costs, they allow both the diagnosis of hemoglobinopathies and identification of its genotype. Additionally, molecular tests allow for the identification of carriers, diagnosis in newborns, and assisting in genetic counseling.
1.3 Prevalence
Worldwide, approximately 5.2% of the population is thought to have at least one genetic alteration in the genes that code for globin with a total of 3.4% affected children with mortality under 5 years. According to forecasts, there are approximately 270 million carriers of variant hemoglobin and 300–400 thousand born with sickle cell anemia or with severe forms of thalassemia (Modell and Darlison 2008; Weatherall 2008). In Africa, 18.2% of the population have hemoglobinopathies of clinical relevance, this a total of 10.68 per 1000 conceptions are affected and results in a 6.4% mortality in children under 5 years old (Modell and Darlison 2008). A study by Piel et al. (2013a, b), on the sickle cell trait, stipulated frequencies between 7.5 and 12.5% in the areas around Lake Victoria, Tanzania, Kenya, and Mozambique. In Mozambique, a study conducted by Willcox et al (1986) found 5–6% of the pregnant women had the sickle cell trait (Hb S) and a case of Hb D. To this day, a high prevalence of sickle cell anemia is expected in the North and Centre of the country due to the high occurrence of malaria in the area (Piel et al. 2010; Gonçalves et al. 2016).
In 2016, the Ministry of Health and the National Institute of Health Statistics reported that 64% of children aged 6–59 months and 54% of women of reproductive age (15–49 years old) have anemia (Ministério da Saúde and Instituto Nacional de Estatística 2016), whether the cause is malnutrition or an underlying disease, such as a hemoglobinopathy, is not specified. Overall, there is a need to conduct studies on the current frequency of hemoglobinopathies in Mozambique and the circulating genotypes. This study aimed to identify hemoglobinopathies in newborns at the Obstetrical Gynecology Department of the Maputo Central Hospital and characterize their electrophoretic patterns and genotypes.
2 Materials and methods
2.1 Study location
The study was performed at the Hospital Central de Maputo (HCM), Gynecology-Obstetrics Department for participants’ enrolment and sample collection, hemoglobin electrophoresis testing was done at the Clinical Analysis Laboratory, and molecular testing was completed at the Biotechnology Centre at Eduardo Mondlane University (CBUEM) laboratory, in Maputo City.
2.2 Sampling
2.2.1 Study population
The study population was newborns at the HCM’s Maternity Ward. Pregnant women that met the selection criteria for the study and signed the informed consent form were chosen. The inclusion criteria were as follows: pregnant women over 17 years old, women capable to provide informed consent, and placenta availability from pregnant women over 34 weeks of gestation with normal delivery or cesarean section. We excluded pregnant women whose fetuses had received an intrauterine transfusion and/or if there was any medical contraindication concerning the procedure.
2.2.2 Sample size
The sample for the study was based on the number of deliveries attended at the Obstetrics and Gynecology Department—HCM. Using the equation from (Daniel and Cross 2013) and a minimum of 242 participants needed was calculated.
2.2.3 Sample collection
In similar studies, umbilical cord blood samples were used to screen for hemoglobinopathies (Mantikou et al. 2010; Rajatileka et al. 2013; Frömmel 2018). The sample was collected from the umbilical cord, after delivery and before the placenta was expelled, the cord was clamped and then cut (Skaar and Renbarger 2012; Rajatileka et al. 2013). The selected area for sample collection was disinfected with gauze and antiseptic solution. The needle was inserted close to the clamped spot and the sample was collected in 10 mL syringes. Afterward, the sample was distributed into anticoagulant tubes containing ethylenediaminetetraacetic acid (4 mL in each tube) and labeled with the mother’s data and the collection date. Then, the samples and forms were transported to the laboratory in cooler boxes and kept at temperatures of 4–8 °C, where after electrophoresis they were sent to CB-UEM for DNA extraction and testing.
2.3 Hemoglobinopathy screening
Hemoglobin separation and quantification were performed using the capillary electrophoresis method using the CAPILLARYS 2 FLEX-PIERCING equipment (Sebia, France). In this technique, the charged molecules are separated by their electrophoretic mobility, in an alkaline buffer with specific pH, the separation occurs according to the pH of the electrolyte and the electroosmotic flow from the anode to the cathode. The plasma is discarded, then the red cells were hemolyzed using a specific solution and injected into the anodic tip of the capillary. Protein separation at high voltages took place in silica capillaries, and hemoglobin fractions were detected at 415 nm at the cathode. The results were analyzed through the program Phoresis Rel v9.2.0 (Sebia, France), which separates the electropherograms into zones, each peak of hemoglobin appears in a specific zone (Mantikou et al. 2010; Frömmel 2018). All samples diagnosed positive for hemoglobinopathies were then genotyped to identify the underlying mutations.
2.4 Molecular analyses
Genomic DNA was extracted using the PureLink® Genomic DNA Kit (Thermo Fisher Scientific, United States of America) following the manufacturer’s protocol “Purification Procedure for Whole Blood” (Life Technologies 2013). Only positive samples for hemoglobinopathies on the capillary electrophoresis (described above) were genotyped using polymerase chain reaction (PCR) techniques and sequencing.
Alpha and hemoglobin variant genotyping was performed using the PCR techniques, both conventional and real-time, gap-PCR. The published protocol by Munkongdee et al. (2010) was adapted to genotype α-thalassemia, and for hemoglobin variants, the protocol published by Vrettou et al. (2003) was adapted. All PCRs were run in parallel with a control sample from an individual with no evidence of any thalassemia or hemoglobin variant.
2.4.1 α-thalassemia real-time PCR
For the genotyping of α-thalassemia, the real-time, gap-PCR technique described by Munkongdee et al. (2010) was used. The primers used were 3′-ψζ-F, 3′-ψζ-R, SEA-R, α2-F, and α2-R published by Munkongdee et al. (2010) for amplification of the HBA2 and HBPZ1 genes (Table 1). Applying Munkongdee et al. (2010) protocol the real-time PCR produced unspecific peaks in the melting curves when doing multiplex PCR. Thus, were decided to run each set of primers separately to identify the specific bands and peaks for each PCR. The real-time PCR test was done using Fast SYBR™ Green Master Mix (Applied Biosystems, ThermoFisher Scientific, USA) with the CFX96 Touch Real-Time PCR Detection System (Bio-Rad, USA). The real-time PCR was optimized for the following parameters: initial denaturation for 5 min at 95 °C, 40 cycles of amplification with the following conditions 94 °C for 30 s, 63 °C for 30 s, 72 °C for 30 s, and the final elongation for 5 min at 72 °C. The melting curves conditions were from 75 to 95 °C at a rate of 0.2 °C per 10 s. The final concentrations for the PCR reagents for HBA2 real-time PCR were 1 × Fast SYBR™ Green Master Mix, 0.2 µM of α2-F and α2-R primers, and 2–4 ng/µL of genomic DNA (gDNA). The HBPZ1 real-time PCR final reagent concentrations: 1 × Fast SYBR™ Green Master Mix, 0.2 µM of the 3′-ψζ-F primers, 0.1 µM of the 3′-ψζ-R and SEA-R primers, and 5–10 ng/µL of gDNA. Samples were sequenced using Sanger sequencing at the company Macrogen Europe (The Netherlands) to confirm the identity of the PCR products.
2.4.2 Hemoglobin variant PCR
The genotyping of hemoglobin variant was done using primers βG-F and βG-R published by Vrettou et al. (2003) that amplify part of the HBB gene (Table 1). The Phusion® High-Fidelity PCR Master Mix with HF Buffer (New England Biolabs, USA) was used. The PCRs were optimized for the following PCR cycling conditions: initial denaturation for 5 min at 95 °C, for amplification it was 35 cycles of 94 °C for 3 s, 60 °C for 5 s, 72 °C for 20 s, and the final elongation for 10 min at 72 °C. The final concentrations of the PCR reagents were 1 × Phusion Master Mix, 0.5 µM of βG-F and βG-F primers, and 2–5 ng/µL gDNA. Samples were then genotyped by Sanger sequencing at the company Macrogen Europe (The Netherlands).
2.5 Statistical analysis
The data were collected using the data collection form and digitalized with Excel (Microsoft Corporation 2018). Statistica 13 v13.3 (TIBCO Software Inc 2017) was used for all statistical analyses. To guarantee the quality of the data, data such as gestational age and number of the participant were verified, to avoid duplication of information, a second observer confirmed the data consistency. The data were summarized in demographic data, characteristics of the participants, and mean melting temperature (Tm). A t test was conducted to compare Hb Bart percentages with genotype.
2.6 Bioinformatics analysis
Geneious® v10.2.6 (Biomatters Ltd 2018) was used for visualization, cleaning, assembly, and analysis of the sequences. The sequences were compared with the reference sequences NG_000007.1 (NCBI 2013; O’Leary et al. 2016) for the β chain gene to identify mutations using the MUSCLE alignment algorithm (Edgar 2004). The identity of sequenced PCR products was confirmed using the Basic Local Alignment Search Tool (BLAST) (Altschul et al. 1990; Morgulis et al. 2008).
3 Results and discussion
During the study period, a total of 2169 parturients were received in the delivery room, 244 were included in the study, of which twelve samples (4.9%) were rejected, and 232 (95.1%) were analyzed. Two hundred twenty-one (221) were Mozambican while eleven were of other nationalities.
Of the newborns included in the study, 117 (51%) were female and 115 (49%) were male. The majority of the parturients were aged between 18 and 30 years old (66.4%) and those over 36 years old were in a lower percentage (10.8%) (Table S1). Gestational ages ranged from 34 to 42 weeks, with pregnant women aged 39 and 40 weeks presenting the highest frequencies 28.9% and 26.7%, respectively. About 80.6% of the parturients were born in Maputo followed by the provinces of Gaza with 4.7% and Sofala with 3.9% (Table S1).
3.1 Hemoglobinopathy screening
Hemoglobin capillary electrophoresis was employed to identify α-thalassemia and/or hemoglobin variants, 64.2% (149 cases) presented normal profiles and 35.8% (83 cases) detected hemoglobinopathies traits (Figs. 1 and 2, Table 2), all were found in newborns at the HCM’s Obstetrics-Gynecology Department. Of all hemoglobinopathies traits detected, 76 were positive for α-thalassemia (33.1%), this is consistent with results found in Ghana, which reported 32.6% of positive cases in children between the age of 6 months and 5 years (Mockenhaupt et al. 2004). In Malawi, a recent study reported a prevalence of 45.7% in newborns for α-thalassemia (Tegha et al. 2021), while Egypt found only 9.7% of neonates were diagnosed with α-thalassemia (Youssry et al. 2018).

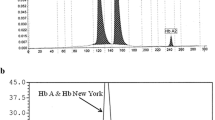
The percentages of the types of hemoglobin found in newborns at the Obstetrical Gynecology Service of the HCM identified using hemoglobin capillary electrophoresis. Hb FA—Fetal and A hemoglobin (normal), Hb Bart—α-thalassemia with Bart’s syndrome, Hb C—hemoglobin C, Hb D—hemoglobin D, and Hb S—sickle cell anemia trait

Electrophoretic profile of the variants found in this study
Our results also show four positives for hemoglobin C (1.6%), two for hemoglobin D (0.8%), and a single sickle-cell case (0.4%). One of the cases of Hb Bart was also diagnosed with Hb C (Hb FA C Bart). Another case was homozygous for Hb D (Hb F D), and other cases were heterozygous for hemoglobin variants (Table 2 and Fig. 2).
Variant hemoglobin are structurally different hemoglobin resulting from nonsynonymous mutations (Loukopoulos 2003; Modell and Darlison 2008). A total of seven newborns were diagnosed with hemoglobin variants, namely Hb S, C, and D, of which the causes are alterations to the HBB gene (Loukopoulos 2003; Thein 2018).
In this study, one case of Hb S, two of Hb D, and four cases of Hb C were discovered using hemoglobin capillary electrophoresis (Table 2 and Fig. 2). A study conducted in 1986 reported a frequency of 5–6% for Hb S and a sole case of Hb D (Willcox et al. 1986). A recent study in the neighboring country of Malawi, reported a prevalence of 0.1% for sickle cell disease and 7.0% of carriers in newborns (Tegha et al. 2021). The difference between this study and that from 1986 was the sampling location and the target group, in the 1986 study samples were from pregnant women mostly from the North and Centre of Mozambique while this study focused on newborns of which the majority are from the South. The only case of Hb S identified was from the North of Mozambique, and according to Willcox et al. (1986) all samples from the south were negative for Hb S. This reinforces the speculation that Hb S is more common in the Centre and North than the South of the country and the presence of other types of circulating hemoglobinopathies in Mozambique, but further studies are needed to confirm focusing on children between the ages of 1 to 5 years.
3.2 Molecular analysis
All eighty-three cases of identified hemoglobinopathies underwent molecular analysis, seventy-seven cases of Hb Bart were tested using a real-time, gap-PCR (named HBA2 PCR and HBPZ1 PCR), while the other six cases of hemoglobin variants were tested using the HBB gene PCR and sent for sequencing.
3.2.1 α-Thalassemia
Regarding molecular analysis, of the seventy-seven α-thalassemia cases, twenty-three were positive to -α3.7/4.2/-α3.7/4.2 (a frequency of 9.9%—Table 3, Fig. 3B and C), and one case where the HBPZ1 gene was not amplified but HBA2 gene was (a frequency of 1.3%—Table 3, Fig. 3D). α thalassemia is usually the result of large deletions on chromosome 16 in the hemoglobin α region (Farashi and Harteveld 2018; Mettananda and Higgs 2018). The twenty-three newborns identified as α3.7/4.2/-α3.7/4.2 corroborated to study on Egyptian neonates in Egypt found, where 9.7% were either heterozygotic or homozygotic for deletions in the HBA2 gene but did not specify which ones (Youssry et al. 2018). The 3.7 kb and 4.2 kb deletions are one of the most common deletions in other countries responsible for causing α-thalassemia (Galanello and Cao 2011; Mettananda and Higgs 2018).

Melting curves of α-thalassemia PCRs. A Melting curve profile of a genotype αα/αα, where one peak around a Tm of 79.4 °C is seen for HBA2 PCR and two peaks for HBPZ1 PCR at Tm 85.8 °C and 80.0 °C are present. B and C Newborns with -α3.7/4.2/-α3.7/4.2 genotype, where no peak or a small peak around 82.5 °C is seen for the HBA2 PCR (Orange) and two peaks for HBPZ1 PCR at Tm 85.8 °C and 80.0 °C (Violet). D A patient with Hb Bart that did not amplify the HBPZ1 gene
The gap-PCR is used to identify large deletions in the α globin gene group and to diagnose α-thalassemia (Liu et al. 2004; Munkongdee et al. 2010). The HBA2 PCR produced a single band at ± 127 bp that produces a peak at 79.4 °C ± 0.2 °C (Fig. 3), which indicates that the newborn is a heterozygous genotype -α3.7/4.2/αα or homozygous αα/αα genotype. This PCR detects the deletions known as 3.7 kb and 4.2 kb (-α3.7/4.2), in the α globin genes cluster region. It should produce a single band of 127 bp with a peak at 85.0 °C if the sample is from an individual with the normal (αα/αα) or heterozygous (-α3.7/4.2/αα) and no amplification for an individual diagnosed with α-thalassemia (-α3.7/4.2/-α3.7/4.2) (Munkongdee et al. 2010). The presence of a smaller peak at 82.5 °C ± 0.2 °C or no peak at all indicates a homozygous genotype -α3.7/4.2/-α3.7/4.2 (Fig. 3).
The HBPZ1 PCR detects the SEA deletion and HBPZ1 gene. The PCR produced two bands one with a peak at 85.8 °C ± 0.4 °C (Fig. 3) with a size of ± 300 bp, another faint band at > 100 bp with a Tm of around 80.8 °C and usually below the threshold (Fig. 3). A band of 307 bp with a peak at 92.8 °C is predicted in samples from individuals of normal phenotype (αα/αα) and a 212 bp band with a peak at 84.8 °C for individuals diagnosed with α-thalassemia with genotype --SEA/--SEA or --SEA/αα. None of the newborns had the SEA deletion. One sample did not amplify the HBPZ1 gene. This may be a case where the deletion occurs in the MCS-R portion of the chromosome, leaving α-globin genes intact (Sollaino et al. 2010; Galanello and Cao 2011). Such an event could produce the profile seen in Fig. 3F.
The other fifty-three newborns are most likely the αα/αα genotype, heterozygote for the -α/αα or --/αα), have another deletion or a non-deletional mutation (αTα/αTα) that causes Hb Bart disease, but is not detectable using this real-time, gap-PCR. Unfortunately, the gap-PCR used in this study does not distinguish between the 3.7 kb and 4.2 kb deletions. Another study will have to be done to clarify this.
A t test (t (75) = 10.77, p = 0.00) revealed that the percentage of Hb Bart in infants with 3.7 kb and/or 4.2 kb deletions (M = 3.66, SD = 1.39) where significantly higher than those of infants that are either heterozygote for the deletion or may have other mutations (M = 0.99, SD = 0.77) (Fig. 4). These findings are supported by other studies that compared Hb Bart percentage in cord blood to genotype (Rugless et al. 2006; Munkongdee et al. 2011). Rugless et al. (2006) found that Hb Bart percentage in cord blood could be used to predict α-thalassemia trait carriers in newborns as well as the associated genotype. Although when comparing the numbers, our data suggest that genotype -α3.7/4.2/-α3.7/4.2 has a mean percentage of 3.66 ± 1.39 (Fig. 4), while Rugless et al. (2006) reported a higher Hb Bart level for the same genotype. With the αα/αα genotype, both studies had comparable results. Munkongdee et al. (2011) presented those with normal genotypes or heterozygotes with only the deletion of one α-globin gene have lower than 1% HB Bart, while 3.6–4.6% Hb Bart indicates the deletion of two α-globin genes. Our research collaborates these results (Fig. 4).

Comparison of Hb Bart percentages between newborns of two different genotypes. Whiskers represent the standard deviation, and the box represents the standard error
3.2.2 Hemoglobin variants
The PCR using primers from Vrettou et al. (2003) amplifies a region of ± 580 bp that includes approximately 150 bp upstream of the HBB gene (nucleotide 70,438),Footnote 1 to amino acid 89 (nucleotide 70,994)1. Therefore, it detects mutations that cause β-thalassemia and those that cause variant hemoglobin such as those identified in this study. A total of seven newborns were identified to have hemoglobin variants, Hb C (A11, A66, A70, and A78), Hb D (A18 and A52), and Hb S (A45) (Figs. 2 and 5).

Alignment of HBB gene amplicon from seven newborns identified of having hemoglobin variants. Y = heterozygote for nucleotides T and C, W = heterozygote for nucleotides A and T
The alignment of the sequences against the reference sequence (NC_000007.1) revealed a point mutation in all samples, a synonymous single nucleotide polymorphism (SNP) at nucleotide 70,6031T to C. This SNP does not alter the amino acid but could be characteristic of the Mozambican population. Significant mutations were observed in newborns A45 and A18 (Fig. 5). The sequencing result revealed that patient A45 (Fig. 5) is a heterozygote for a nonsynonymous mutation at nucleotide 70,6141 A to T, converting the β6 from glutamic acid (Glu/E) to valine (Val/V) (Fig. 5). This mutation is characterized as the cause of the production of Hb S, sickle-cell disease (Thein 2018; Williams and Thein 2018).
Unfortunately, this primer set does not cover the full HBB gene, and thus amino acid β121, which is important in genotyping individuals with Hb D, is not included (Hardison et al. 1998; Loukopoulos 2003). Individuals with the Glu to glutamine (Gln) mutation at β121, are usually carriers of the Hb D. Newborn sample A18 (Fig. 5) had a mutation at nucleotide 70,866 G > T, resulting in a change of β47 aspartic acid (Asp) to tyrosine (Tyr). While it is not the residue or the mutation associated with Hb D, it is a hemoglobin variant discovered in a Mozambican child in 1983, known as Hb Maputo (Marinucci et al. 1983; Hardison et al. 1998). This does not exclude the possibility that this newborn is a carrier of two types of Hb variants, Hb Maputo and Hb D. Newborn A52 (Fig. 5), also identified as a carrier of the Hb D variant, presented no other mutations. Therefore, to confirm their genotype for Hb D mutation at amino acid β121 amplification of the rest of the gene is required.
Carriers of the Hb C variant typically have a mutation at the β6 Glu to lysine (Lys) (Hardison et al. 1998; Loukopoulos 2003), in the four newborns (A11, A66, A70, and A78—Fig. 5) no mutations were identified, not even as heterozygotes. Considering that all the newborns had Hb C percentages below 1.0, it could have been a false positive. Thus, these newborns would have to be retested. Hemoglobin C is more dominant in West Africa (Piel, et al. 2013a, b); therefore, it is most likely that it was either an error in the screening using hemoglobin capillary electrophoresis or contamination from the mother’s blood when collecting the sample (Morin et al. 2017).
The above results highlight the need to further investigate hemoglobin variants within Mozambique. According to Williams and Thein (2018), since malaria is endemic to Mozambique there is a high frequency of sickle-cell traits within the population. Our study, consisting of newborns predominantly from southern Mozambique, revealed an incredibly low prevalence of 0.4% of Hb S but detected the presence of other Hb variants. It could be that, based on previous studies (Willcox et al. 1986; Piel et al. 2013b), if we focus on the center and northern part of the country the frequency of Hb S will be higher. Concerning the other hemoglobin variants, Hb C and Hb D are the third and fourth most common hemoglobin variants of clinical significance (Piel et al. 2013b; Torres et al. 2015). In our study, we have a prevalence of 0.8% for Hb D, one of which had a mutation identified as the Hb Maputo variant. To confirm the genotype of both newborns the full HBB needs to be sequenced. Thus, future studies should use a more inclusive genotyping tool.
4 Conclusion
Our study confirmed the presence of α-thalassemia and hemoglobin variants of clinical importance in the Mozambique population, to what degree this impacts public health and its distribution throughout the country remains to be explored.
With α-thalassemia, we identified the 3.7 kb and/or 4.2 kb deletions of α-globin genes within our population, with a 9.9% frequency. On top of that, there may be other deletions that have not been detected by the molecular test we used and there is evidence of a deletion in the 3’region of the HBZP1 gene that needs to be characterized. Furthermore, we did not evaluate for non-deletional mutations in the α-globin gene cluster. This could be a cause for concern due to the risk of severe forms of thalassemia and thus further investigation and monitoring are required. What is clear is that we have only scratched the surface in characterizing α-thalassemia and the underlying genotypes associated with it in the Mozambican population. The next steps will be finding a more comprehensive genotyping tool to detect a broader range of deletions and as well as non-deletional mutations in the α-globin gene cluster.
This study confirmed the presence, although at low levels, of other Hb variants, namely Hb S, Hb D, and Hb Maputo in our population. Further studies are needed to understand the distribution of variants within Mozambique (South to North) and, future projects should also include the screening of individuals after 1 year to identify β-thalassemia cases. As with the α-globin gene cluster, a broader molecular test is required to genotype not only Hb variants (i.e., the full HBB gene) but also point mutations and deletions in the 5’ UTR that may result in β-thalassemia.
As a pilot study on the status of hemoglobinopathies and their associated genotypes in Mozambique, we believe that critical genotypes for α-thalassemia were identified and the confirmed presence of clinically significant hemoglobin variants. These results reinforce the importance and need of both electrophoresis and genetic screening in both newborns and older patients to provide a better healthcare service. Still, a more comprehensive study that will better evaluate the status of hemoglobinopathies and their circulating genotypes nationwide is needed. Despite its limitations, this study can serve as a basis for future research.
Availability of data and material (data transparency)
Participants of this study did not agree for their data to be shared publicly; therefore, supporting raw data are not available.
Code availability
Not applicable.
Abbreviations
- Hb:
-
Hemoglobin
- HBA1:
-
Hemoglobin subunit alpha 1
- HBA2:
-
Hemoglobin subunit alpha 2
- HBB:
-
Hemoglobin subunit beta
- HBPZ1:
-
Hemoglobin subunit zeta pseudogene 1
- HPLC:
-
High-performance liquid chromatography
- IEF:
-
Isoelectric focusing
- SEA:
-
Southeast Asian deletion
References
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Benson JM, Therrell BL (2010) History and current status of newborn screening for hemoglobinopathies. Semin Perinatol 34:134–144. https://doi.org/10.1053/j.semperi.2009.12.006
Biomatters Ltd (2018) Geneious (10.2.6). https://www.geneious.com
Daniel WW, Cross CL (2013) Biostatistics: a foundation of analysis in the health sciences, 10th edn. Wiley, Hoboken
de Souza Torres L, Okumura JV, da Silva DGH, Bonini-Domingos CR (2015) Hemoglobin D-Punjab: origin, distribution and laboratory diagnosis. Rev Bras Hematol Hemoter 37:120–126. https://doi.org/10.1016/j.bjhh.2015.02.007
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Farashi S, Harteveld CL (2018) Molecular basis of α-thalassemia. Blood Cells Mol Dis 70:43–53. https://doi.org/10.1016/j.bcmd.2017.09.004
Frömmel C (2018) Newborn screening for sickle cell disease and other hemoglobinopathies: a short review on classical laboratory methods—isoelectric focusing, HPLC, and capillary electrophoresis. Int J Neonatal Screen 4:39. https://doi.org/10.3390/ijns4040039
Galanello R, Cao A (2011) Alpha-thalassemia. Genet Med 13:83–88. https://doi.org/10.1097/GIM.0b013e3181fcb468
Gonçalves BP, Gupta S, Penman BS (2016) Sickle haemoglobin, haemoglobin C and malaria mortality feedbacks. Malar J 15:1–7. https://doi.org/10.1186/s12936-015-1077-5
Goonasekera HW, Paththinige CS, Dissanayake VHW (2018) Population screening for hemoglobinopathies. Annu Rev Genomics Hum Genet 19:355–380. https://doi.org/10.1146/annurev-genom-091416-035451
Hardison RC, Chui DHK, Riemer CR et al (1998) Access to a syllabus of human hemoglobin variants (1996) via the world wide web. Hemoglobin 22:113–127. https://doi.org/10.3109/03630269809092136
Life Technologies (2013) PureLink ® Genomic DNA Kits For purification of genomic DNA. PureLink Genomic DNA Kits For purification of genomic DNA 1–48
Liu JZ, Ou CY, Wang LR et al (2004) Detection of three common, deletional α-thalassemia determinants in southern china by a single-tube multiplex polymerase chain reaction method. Hemoglobin 28:39–44. https://doi.org/10.1081/HEM-120028885
Liu J, Yan M, Wang Z et al (2006) Molecular diagnosis of α-thalassemia by combining real-time PCR with SYBR Green1 and dissociation curve analysis. Transl Res 148:6–12. https://doi.org/10.1016/j.lab.2006.03.016
Loukopoulos DL (2003) Haemoglobinopathies. In: Encyclopedia of Life Sciences. Wiley, Chichester, pp 1–10
Mantikou E, Harteveld CL, Giordano PC (2010) Newborn screening for hemoglobinopathies using capillary electrophoresis technology: testing the Capillarys® Neonat Fast Hb device. Clin Biochem 43:1345–1350. https://doi.org/10.1016/j.clinbiochem.2010.06.009
Marinucci M, Boissel JP, Massa A et al (1983) Hemoglobin maputo: a new 6-chain variant (α 2 β 2 47 (Cd6) Asp→Tyr) in combination with hemoglobin S, identified by high performance liquid chromatography (Hplc). Hemoglobin 7:423–433. https://doi.org/10.3109/03630268309038411
Mettananda S, Higgs DR (2018) Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin North Am 32:177–191. https://doi.org/10.1016/j.hoc.2017.11.003
Microsoft Corporation (2018) Microsoft Excel. https://office.microsoft.com/excel
Ministério da Saúde, Instituto Nacional de Estatística (2016) Inquérito de Indicadores de Imunização, Malária e HIV/SIDA em Moçambique (IMASIDA) 2015. Maputo
Mockenhaupt FP, Ehrhardt S, Gellert S et al (2004) α+-thalassemia protects African children from severe malaria. Blood 104:2003–2006. https://doi.org/10.1182/blood-2003-11-4090
Modell B, Darlison M (2008) Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 86:480–487. https://doi.org/10.2471/BLT.06.036673
Morgulis A, Coulouris G, Raytselis Y et al (2008) Database indexing for production MegaBLAST searches. Bioinform Original Paper 24:1757–1764. https://doi.org/10.1093/bioinformatics/btn322
Morin AM, Gatev E, McEwen LM et al (2017) Maternal blood contamination of collected cord blood can be identified using DNA methylation at three CpGs. Clin Epigenetics 9:75. https://doi.org/10.1186/s13148-017-0370-2
Munkongdee T, Vattanaviboon P, Thummarati P et al (2010) Rapid diagnosis of α-thalassemia by melting curve analysis. J Mol Diagn 12:354–358. https://doi.org/10.2353/jmoldx.2010.090136
Munkongdee T, Pichanun D, Butthep P et al (2011) Quantitative analysis of Hb Bart’s in cord blood by capillary electrophoresis system. Ann Hematol 90:741–746. https://doi.org/10.1007/s00277-010-1137-4
NCBI (2013) National Center for Biotechnology Information (NCBI). http://www.ncbi.nlm.nih.gov/. Accessed 21 Feb 2013
O’Leary NA, Wright MW, Brister JR et al (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44:D733–D745. https://doi.org/10.1093/nar/gkv1189
Piel FB, Patil AP, Howes RE et al (2010) Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun 1:104. https://doi.org/10.1038/ncomms1104
Piel FB, Howes RE, Patil AP et al (2013a) The distribution of haemoglobin C and its prevalence in newborns in Africa. Sci Rep 3:1–8. https://doi.org/10.1038/srep01671
Piel FB, Patil AP, Howes RE et al (2013b) Global epidemiology of Sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 381:142–151. https://doi.org/10.1016/S0140-6736(12)61229-X
Rajatileka S, Luyt K, El-Bokle M et al (2013) Isolation of human genomic DNA for genetic analysis from premature neonates: a comparison between newborn dried blood spots, whole blood and umbilical cord tissue. BMC Genet 14:1–9. https://doi.org/10.1186/1471-2156-14-105
Rugless MJ, Fisher CA, Stephens AD et al (2006) Hb Bart’s in cord blood: an accurate indicator of α-thalassemia. Hemoglobin 30:57–62. https://doi.org/10.1080/03630260500454550
Sabath DE (2017) Molecular diagnosis of thalassemias and hemoglobinopathies: an ACLPS critical review. Am J Clin Pathol 148:6–15. https://doi.org/10.1093/AJCP/AQX047
Skaar C, Renbarger JL (2012) Comparison Between Umbilical Cord Blood and Buccal Swabs. 204(4):1–10. https://doi.org/10.1016/j.ajog.2010.12.013.Collection
Sollaino MC, Paglietti ME, Loi D et al (2010) Homozygous deletion of the major alpha-globin regulatory element (MCS-R2) responsible for a severe case of hemoglobin H disease. Blood 116:2193–2194. https://doi.org/10.1182/blood-2010-04-281345
Tegha G, Topazian HM, Kamthunzi P et al (2021) Prospective newborn screening for sickle cell disease and other inherited blood disorders in Central Malawi. Int J Public Health 66:629338. https://doi.org/10.3389/ijph.2021.629338
Thein SL (2018) Molecular basis of β thalassemia and potential therapeutic targets. Blood Cells Mol Dis 70:54–65. https://doi.org/10.1016/j.bcmd.2017.06.001
TIBCO Software Inc (2017) Statistica TM v13.3 (13.3; Issue June). TIBCO Software Inc
Vrettou C, Traeger-Synodinos J, Tzetis M et al (2003) Rapid screening of multiple β-globin gene mutations by real-time PCR on the LightCycler: application to carrier screening and prenatal diagnosis of thalassemia syndromes. Clin Chem 49:769–776. https://doi.org/10.1373/49.5.769
Weatherall D (2008) Hemoglobinopathies worldwide: present and future. Curr Mol Med 8:592–599. https://doi.org/10.2174/156652408786241375
Willcox MC, Liljestrand J, Bergstrom S (1986) Abnormal haemoglobins among pregnant women from Mozambique. J Med Genet 23:151–152. https://doi.org/10.1136/jmg.23.2.151
Williams TN, Thein SL (2018) Sickle cell anemia and its phenotypes. Annu Rev Genomics Hum Genet 19:113–147. https://doi.org/10.1146/annurev-genom-083117-021320
Youssry I, el Badawy A, Samy RM et al (2018) Prevalence of α-thalassemia in the Egyptian population. Hemoglobin 42:243–246. https://doi.org/10.1080/03630269.2018.1527231
Acknowledgements
The authors address thanks to the nurses, doctors, and technical personnel from the Department of Gynecology and Obstetrics, the Clinical Analysis Laboratory, and the Medical Genetics Service of the Maputo Central Hospital, for their constant support in the collection and processing of samples.
Funding
Training in biodiversity and biotechnology—Eduardo Mondlane University AID 11096 from the Italian Agency for Development Cooperation—Maputo (Agenzia Italiana per la Cooperazione allo Sviluppo—Maputo). Bilateral Research Program UEM-SWEDEN 2017–2022.
Author information
Authors and Affiliations
Contributions
DRAB carried out the molecular analysis of samples, data analysis, and took the lead in writing the manuscript. JM carried out sampling, screening of samples, and contributed to the writing of this manuscript. All authors contributed to the conception of the project, provided critical feedback, and gave final approval for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest.
Ethics approval
The protocol was approved by the National Health Bioethics Committee of the Ministry of Health of Mozambique, reference number: 409/CNBS/18.
Consent to participate
All participants included in this study gave their informed consent.
Consent for publication
All participants gave their consent for publication of the results of this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This paper belongs to a Topical Collection originated from a long scientific collaboration between Mozambican and Italian universities promoted by the Italian Agency for Development Cooperation.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Brito, D.R.A., Mutchamua, J.R., Chilundo, B. et al. Electrophoretic and molecular profile of the hemoglobinopathies in newborns at a reference hospital in Mozambique. Rend. Fis. Acc. Lincei 33, 283–293 (2022). https://doi.org/10.1007/s12210-022-01073-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12210-022-01073-w