
Abstract
In fast muscle, isoforms of troponin T (TnT) contain an N-terminal hypervariable region that does not bind any protein of the thin filament. The N-terminal domain of TnT is removed by calpain during stress conditions and so could modulate the role of TnT in the regulation of contraction by affecting the TnT-binding affinity for tropomyosin (Tm) depending on the sequence and charge within the domain. During skeletal muscle contraction, the myokinase reaction is displaced by AMP deaminase (AMPD), an allosteric metalloenzyme, toward the formation of ATP. An unrestrained AMPD activity follows the proteolytic cleavage of the enzyme in vivo that releases a 97 aa N-terminal fragment, removing the inhibition exerted by the binding of ATP to a zinc site in the N-terminal region. Rabbit fast TnT or its phosphorylated 50-aa residue N-terminal peptide restores in AMPD the inhibition by ATP, removed in vitro by the release of a 95 aa N-terminal fragment by trypsin. Since the N-terminal region of fast rabbit TnT contains a putative zinc-binding motif, it can be inferred that TnT mimics the regulatory action exerted in native AMPD by the N-terminal domain that holds the enzyme in a less active conformation due to the presence of a zinc ion connecting the N-terminal and C-terminal regions. Together with evidence that AMPD is localized on the myofibril, the data reported in this review on the interactions between AMPD and TnT strongly suggest that these proteins mutually combine to fine-tune the regulation of muscle contraction in fast muscle.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Troponin (Tn) is a complex of three proteins, namely troponin I (TnI), troponin C (TnC) and troponin T (TnT), that is bound to tropomyosin (Tm) in the thin filament and regulates the contractile activity of striated myofibrils. As Ca2+ binds to TnC, a cascade of conformational changes occurs in Tn that causes the movement of Tm and the release of the TnI inhibition on the actomyosin ATPase, exposing the myosin-binding sites on F-actin subunits and allowing cross-bridge cycling and fiber contraction (Gordon et al. 2000).
While the C-terminal domains of fast, slow and cardiac TnT are conserved and their role in anchoring the Tn complex to Tm on the thin filament is well established (Perry 1998; Gomes et al. 2002), to date no clear function has been envisaged for the N-terminal region that shows a high degree of variability depending on the muscle type and development stage (Medford et al. 1984; Breitbart et al. 1985; Jin et al. 1992; Jin 2016). However, there is evidence that the TnT N-terminal domain significantly affects the TnT-binding affinity to Tm depending on its sequence and charge (Amarasinghe and Jin 2015). This observation could form the molecular basis for the hypothesized role of TnT as modulator of Tn Ca2+ sensitivity and force production (Gomes et al. 2004). Actually, TnT has been found to activate the Mg ATPase of actomyosin in the presence of Tm, but without the other components of the troponin complex (Malnic et al. 1998).
While to date no protein that could bind in vivo the TnT N-terminal elongated region has been identified, the data of the literature give evidence of the in vitro interaction of rabbit fast-twitch TnT and of its N-terminal domain with AMP deaminase (AMPD), a strictly regulated zinc metalloenzyme that is highly expressed in white muscle. The N-terminal region of rabbit skeletal muscle AMPD contains a zinc-binding site that plays a critical role in preserving the conformation of the catalytic dinuclear zinc center that is involved in the modulation of the enzyme activity (Mangani et al. 2007; Martini et al. 2007). During muscle strenuous exercise, upon removal of the N-terminal region by a calpain-like protease, AMPD is no longer inhibited by ATP and the ensuing enzyme activation is responsible for the observed large muscle ammonia accumulation (Martini et al. 2004). Based on the observations summarized in this paper that the N-terminal domain of rabbit fast TnT contains a putative zinc-binding motif and that the addition of native TnT or the phosphorylated N-terminal peptide of TnT restores the allosteric properties of native AMPD that were removed by the limited proteolysis, i.e., inhibition by ATP, we hypothesize that, following the proteolytic cleavage of the enzyme during strenuous exercise, the unrestrained AMPD could revert to the less active native conformation, possibly through the interaction of the catalytic Zn ion present at the C-terminus of the enzyme with the putative metal-binding site found at the N-terminal regulatory region of TnT.
In summary, we suggest that among the physiological roles envisaged for TnT, the interaction in vivo of its N-terminal region with AMPD should be taken in consideration as a physiological mechanism that switches off the activated enzyme to allow the recovery of the adenylate pools after intense muscle contraction.
2 Troponin T and the calcium sensitivity of the MgATPase of actomyosin
Contraction of striated (i.e., cardiac and skeletal) muscle is regulated via the thin (actin) filament by the concerted efforts of Tm and the Tn complex. It is generally accepted that in the resting state, the myofibrillar actomyosin MgATPase of striated muscle is inhibited by the blocking of the interaction of actin with myosin, although some of the molecular details of this mechanism are as yet unclear.
It is well known that TnI is the component of the troponin complex that inhibits the MgATPase of actomyosin (Perry 1999). Calcium is required for TnI to switch from the inhibitory to the non-inhibitory mode—the so-called calcium sensitivity of the MgATPase of actomyosin. Using actomyosin preparations, it has been observed that TnC neutralizes the inhibitory activity of TnI even in the absence of calcium, but sensitivity to calcium is restored when the whole troponin complex is present (Amphlett et al. 1976). These results indicate that the calcium-induced switch of the troponin complex on the thin filament is promoted by TnT, the Tm-binding subunit of the Tn complex that presumably lowers the affinity of TnC for TnI (Potter et al. 1995).
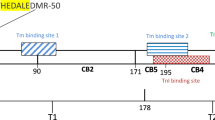
The functional domains of skeletal muscle TnT (Fig. 1) have been identified using fragments of TnT obtained by limited chymotryptic and CNBr digestions (Ohtsuki 1979; Jackson et al. 1975; Pearlstone et al. 1977; Pearlstone and Smillie 1977, 1981) (Fig. 1). Both chymotryptic fragments T1 and T2 bind Tm (Ohtsuki 1979). Although it had been shown that CNBr fragment CB2 [comprising amino acid (aa) residues 91–171 of rabbit skeletal muscle TnT] binds to tropomyosin less strongly than the TnT CNBr fragment CB1 (aa residues 1–171) (Pearlstone and Smillie 1982), it was subsequently reported that TnT46–259 produced by endogenous muscle protease activity has the same affinity (Ohtsuki et al. 1984) or even a greater affinity (Pan et al. 1991) than the intact protein for Tm. More recent studies using genetically engineered TnT fragments (Jin and Chong 2010) confirmed that the Tm-binding site in the T1 region lies inside a region that corresponds to aa residues 79–117 of the longest rabbit fast skeletal muscle TnT variant, in agreement with the previous observation that the CNBr peptide CB2 forms a complex with Tm (Jackson et al. 1975). The Tm-binding site located in the T2 region encompasses the aa residues 179–218 (Jin and Chong 2010).

Schematic ribbon diagram representing the primary structure and the functional domains of Troponin T from rabbit fast skeletal muscle. The residue numbers are those of the 279 aa long rabbit fast TnT (accession number P02641.4). The scheme indicates the positions of the CNBr peptides (CB) (Jackson et al. 1975; Pearlstone et al. 1977). CB1, not shown, is a partial digestion product comprising CB3 and CB2. T1 and T2 are the chymotryptic fragments of rabbit fast TnT (Ohtsuki 1979). The Tm-binding sites 1 (79–117 region) and 2 (179–218 region) (Jin and Chong 2010) are the Tm-binding sites located in the T1 and T2 fragments, respectively. The region 244–257 corresponds to the human cardiac TnC-binding site (Takeda et al. 2003) and the region 190–235 corresponds to the chicken fast skeletal muscle TnI-binding site (Vinogradova et al. 2005) when the rabbit fast TnT sequence is aligned with those from the two different species using Clustal Omega. The 29–50 region corresponds to the putative metal-binding site at the N-terminal variable region of rabbit fast TnT.  represents the position of ser 1 residue that can be phosphorylated by phosphorylase b kinase and TnT kinase (Moir et al. 1977; Dobroval’skii et al. 1976)
represents the position of ser 1 residue that can be phosphorylated by phosphorylase b kinase and TnT kinase (Moir et al. 1977; Dobroval’skii et al. 1976)
Even though the aa sequences of the two Tm-binding sites of fast, slow and cardiac TnT isoforms are conserved in avian and mammalian species (Jin and Chong 2010), major differences have been reported in the literature regarding the binding of TnT isoforms to Tm: in rabbit fast skeletal muscle, fragments T1 and T2 have comparably high affinity for Tm (Heeley et al. 1987), while mouse slow skeletal muscle T1 binds to Tm with an affinity much lower than that shown by T2 (Jin and Chong 2010). These discrepancies have been attributed to the hypervariability of the N-terminal T1 region of the different muscle TnT isoforms (Wei and Jin 2011, 2016) that might affect the affinity of the Tm-binding sites. A functional role of the N-terminus of TnT is suggested by the observation that its structure is regulated by alternative splicing during postnatal development of skeletal muscle with the production of variants that alter the length and acidity of the N-terminal domain (Perry 1998).
The strongly acidic N-terminal domain of TnT probably adopts an extended and accessible conformation, since ser1 is readily phosphorylated by TnT kinase (Dobroval’skii et al. 1976) and phosphorylase b kinase (Moir et al. 1977). While functional differences are found between intact TnT isoforms, deletion of the N-terminal region does not eliminate the TnT activity in the Ca2+ activation of actomyosin ATPase (Pearlstone and Smillie 1982; Ohtsuki et al. 1984), indicating that the N-terminal variable region is not essential for TnT’s core activity in the regulation of muscle contraction. Moreover, the CB3 fragment encompassing residues 1–90 in rabbit fast skeletal TnT represents a hypervariable region of the protein that does not bind any known thin filament protein (Perry 2001). However, a special role in the calcium switch has been envisaged for this region of TnT on the basis of the observation that chicken skeletal muscle TnT1–191, i.e., a fragment derived from a single troponin subunit, activates the MgATPase of actomyosin in the presence of Tm to levels observed for the whole Tn complex in the presence of calcium (Malnic et al. 1998). It had previously been reported that removal of the N-terminal 38 residues of bovine cardiac TnT by chemical cleavage (TnT39–284) lowered the calcium sensitivity of acto-S1 ATPase (Tobacman 1988). The hypothesis that the structural variations of the N-terminus region might induce secondary conformational changes in other domains of TnT has also been suggested by Amarasinghe and Jin (2015) who have shown that the T1 fragment obtained from chicken breast muscle TnT, which contains an unusually long and charged N-terminal region, binds with a lower affinity to a Tm-affinity column compared to the recombinant T1 fragment of rabbit fast TnT, while no differences in affinity for Tm were observed for the C-terminal Tm-binding site within the T2 fragment of fast TnT from the two species. These data were further corroborated by the observation that the deletion of the N-terminus of the T1 fragment abolishes the differences of fast, slow and cardiac T1 recombinant fragments in the binding affinity for Tm, indicating that the N-terminal region exerts a modulatory role on reducing the affinity for Tm.
It has been clear from a number of studies that TnC and TnI (Pearlstone and Smillie 1978; Tanokura et al. 1983) are anchored in the C-terminal region of TnT. By using deletion mutants, Malnic et al. (1998) mapped the binding of TnI to the C-terminal region of chicken fast muscle (aa 216–263). More recent crystallization analyses localized the TnI-binding site on the region 200–245 of chicken fast skeletal muscle TnT, corresponding to aa 190–235 in rabbit fast skeletal muscle TnT as indicated in Fig. 1 (Vinogradova et al. 2005) and the binding of TnC was mapped to the 256–270 region of human cardiac TnT, corresponding to aa 244–258 in rabbit fast skeletal muscle TnT (Fig. 1) (Takeda et al. 2003). However, the crystallography data only determined the structure for a portion of the TnT C-terminal region in the Tn complex.
In addition to the ascertained interaction of the TnT T2 region with TnI and TnC, there is some evidence that the N-terminal hypervariable region of the various TnT isoforms might contain sites of contact for TnC (Moir et al. 1977; Tanokura et al. 1983) and for TnI (Raggi et al. 1989).
The chimera proteins composed of an acidic or a basic chicken fast muscle T1 and the mouse cardiac T2 showed that the charge variation of the N-terminus region indeed affected the binding to Tm and TnI, suggesting that the TnT N-terminus region, whose primary sequence varies along with the development of the different muscle types, might modulate the regulatory role of TnT in the fiber contractility (Biesiadecki et al. 2007).
3 AMP deaminase, a very active regulated enzyme of white skeletal muscle
During the various phases of cell metabolism, the adenine nucleotide pool, i.e., the sum of ATP, ADP and AMP concentrations, remains relatively constant; however, the relative concentrations of the adenine nucleotides, represented by the energy charge (Atkinson 1968), is determined by the energy status of the cell and a high value of energy charge is essential for the various forms of cellular work.
When utilization of ATP is kept low, a constant energy charge value is assured by the equilibrium of the adenylate kinase (AK) reaction (2ADP = ATP + AMP). Under conditions of impaired energy metabolism, such as during sustained contractile activity of skeletal muscle that occurs with a rapid utilization of ATP, recovery of normal values of energy charge might be achieved by shifting the equilibrium of the AK reaction toward the production of ATP through the acceleration of the breakdown of AMP that could take place by two different mechanisms, either deamination to IMP or dephosphorylation to adenosine (Fig. 2a). The distribution data in various animal tissues of the two enzymes catalyzing the breakdown of AMP show that in skeletal muscle the AMP deaminase (AMPD) activity is very high as compared to all other tissues, including heart and smooth muscle (Conway and Cooke 1939). While in the heart the two enzymes potentially have similar levels of activity, in white skeletal muscle the catabolism of AMP operates almost exclusively via AMPD (Rubio et al. 1973; Bockman and McKenzie 1979).

Pathways of adenylate metabolism in exercising white skeletal muscle
AMPD is a multimeric enzyme that was first isolated by Schmidt (1928). Extensive work carried out by Parnas and his school well established that ammonia formed by muscle during work arises from deamination of AMP to IMP catalyzed by AMPD (Parnas 1929; Parnas et al. 1930). Parnas considered irreversible the deamination processing of AMP, which could occur during anaerobic contraction, and hypothesized the existence of a regenerative process of AMP from IMP occurring during periods of oxidative recovery (Parnas and Lewinski 1935). The results of subsequent work have fully confirmed this hypothesis (Newton and Perry 1960; Davey 1961). Lowenstein and Tornheim (1971) gave evidence for the occurrence in muscle of a cyclical process, termed the purine nucleotide cycle, consisting of the reactions catalyzed by AMPD, adenylosuccinate synthetase and adenylosuccinase (Fig. 2b) that plays a major role in the reamination of IMP during recovery, when the concentration of the total purine nucleotides slowly reaches the resting values (Lowenstein 1972; Westra et al. 1982). It should be noted that in this context, the deamination of AMP to IMP by AMPD reduces the formation of adenosine, a nucleoside that could leak out of the cell (Rubio et al. 1973), thereby depleting the muscle fiber of the substrate that is required to restore AMP and ATP concentrations during recovery. Nonetheless, there is no doubt that the main function of the removal of AMP by deamination is to favor the AK reaction toward ATP production through the utilization of β-phosphate of ADP in the phases of rapid utilization of ATP by myosin ATPase.
Enzyme distribution data have shown that white muscle from different species catalyzes the deamination of AMP four to ten times more effectively than red muscle that showed about 15% of the AK activity of white muscle (Raggi et al. 1969). It has also been shown that AMPD exists in striated muscle as two different isoenzymes (Raggi et al. 1975; Raggi and Ranieri-Raggi 1987; Barsacchi et al. 1979). In red muscle, the prevalent form (form A) has the chromatographic properties of the single form present in the heart, while the minor form of AMPD found in red muscle corresponds to that which accounts for the greatly increased AMPD activity of white muscle (form B) (Raggi et al. 1975). At the optimal pH value 6.5, both enzymes show hyperbolic substrate–velocity curves and are inhibited by GTP, inducing sigmoid kinetics. An effect similar to that of GTP is exerted on form B by ATP, while form A is almost insensitive to this nucleotide. At pH 7.1, both enzymes follow sigmoid kinetics. ATP enhances the sigmoidicity of the substrate–velocity curve of form B, but it stimulates form A, reverting sigmoidal to hyperbolic kinetics shown by the enzyme at optimal pH (Raggi and Ranieri-Raggi 1987). According to these data, during moderate contractile activity of white skeletal muscle, AMPD is inhibited by ATP (Fig. 2a) and is activated only during the performance of exhaustive work, when a drop of the ATP level occurs and the increase of ADP concentration together with the acidification of tissue due to the production of lactate cancel the action of all inhibitors (Fig. 2b). Interestingly, as reported for the N-terminus of fast skeletal TnT (Wang and Jin 1997), a transition was observed by Kendrick-Jones and Perry (1967) also for AMPD, as well as AK, creatine phosphokinase (CK) and fructose 1,6-diphosphate aldolase, whose activities increase in skeletal muscle during development from fetal to adult type; the adult skeletal muscle, differently from other tissues, showed a more highly developed anaerobic metabolism than its fetal counterpart (Hartshorne and Perry 1962). Three distinct AMPD isoforms are sequentially expressed during the development in rat. The embryonal muscles express the AMPD isoform that is specific of adult non-muscle tissues such as spleen, lung, thymus, blood and liver. Near birth and for the first 2–3 week of life, a different perinatal AMPD isoform is found only in skeletal muscle together with the adult skeletal muscle AMPD isoform that reaches the maximum expression level after 3 week of development (Marquetant et al. 1987).
4 AMPD is a zinc metalloenzyme and HPRG is the subunit that functions as a metallochaperone
The characterization of skeletal muscle AMPD as a zinc metalloenzyme was reported for the rat enzyme (Raggi et al. 1970) as well as for the rabbit enzyme (Zielke and Suelter 1971) on the basis of its interaction with chelating agents and metal ions. Atomic absorption analysis established the presence of 2.0 and 2.6 mol of zinc, respectively, per mole of rat enzyme (mol wt. 290 kDa) and rabbit enzyme (mol wt 278 kDa) (Raggi et al. 1970; Zielke and Suelter 1971). The problem of assigning a precise stoichiometry for the zinc binding to rabbit skeletal muscle AMPD is complicated by the observation that the apoenzyme binds 4 mol of zinc per mol, but the increase of Vmax due to the addition of the fourth zinc atom is only 28% of that expected. This suggested that the fourth zinc atom is not directly associated with activity (Zielke and Suelter 1971).
More recently, Merkler and Schramm (1993) reported one zinc atom content for the 80 kDa subunit of a form of AMPD from baker’s yeast that lacks an N-terminal segment of 192 amino acids as a consequence of a proteolytic cleavage that occurs during purification and proposed the model of a penta-coordinated zinc bound at the catalytic site, since the enzyme showed conservation of the four amino acid residues (three His and one Asp) known from the X-ray crystal structure of adenosine deaminase to bind zinc in contact with the attacking water nucleophile (Wilson et al. 1991). Alignment of the amino acid residues supposed to be in contact with zinc in yeast AMPD with the rabbit fast skeletal muscle enzyme (NCBI accession number: XP_002715794.3) demonstrates conservation of only three amino acid residues (His-363, His-572 and Asp-649, corresponding, respectively, to His-422, His-630 and Asp-707 in yeast AMPD), while His-424 of the yeast enzyme is replaced by Gly-365. Moreover, X-ray absorption spectroscopy of Zn-peptide binary and ternary complexes prepared using a number of synthetic peptides mimicking the potential metal-binding sites of rabbit skeletal muscle AMPD strongly suggested that the region 48–61 of the enzyme contains a zinc-binding site, while region 360–372 is not able to form 1:1 complexes with zinc, in contrast to what has been suggested for the corresponding region of yeast AMPD (Mangani et al. 2007). X-ray absorption spectroscopy performed on fresh preparations of rabbit skeletal muscle AMPD provided evidence for a dinuclear zinc site in the enzyme compatible with a (µ-aqua)(µ-carboxylato)dizinc(II) core with an average of two histidine residues at each metal site and a Zn–Zn distance of about 3.3 Å (Martini et al. 2007). The data also indicated that two zinc ions are bound to the catalytic subunit of the enzyme, one to the three conserved amino acid residues among those four assumed to be in contact with zinc in yeast AMPD, and the other at the N-terminal region, probably to His-51, Glu-53 and His-57 (Fig. 3).

The proposed interactions of the substrate at the dinuclear cocatalytic Zn site of rabbit skeletal muscle AMPD (Mangani et al. 2007; Martini et al. 2007). The amino acid residues involved in the binding in the catalytic Zn1 region are based on the proposed interactions of the substrate transition state at the catalytic site of adenosine deaminase (Wilson et al. 1991) and amino acid homology to rabbit AMPD1
Justification for a structurally bridged dinuclear metallocenter in rabbit skeletal muscle AMPD was supported by N-terminal analysis of the peptides liberated by limited tryptic digestion of different enzyme preparations suggesting the existence of two different protein conformations which is consistent with the hypothesis of the presence of a zinc ion connecting the N-terminal and C-terminal regions of AMPD (Mangani et al. 2007). The hypothesis of a conformational change of AMPD induced or stabilized by zinc binding to the 51–60-residue region (HHEMQAHILH) is strengthened by the observation that carbethoxylation by diethyl pyrocarbonate of one or two histidine residues per subunit results in conversion of rabbit skeletal muscle AMPD into a species that, unlike the native enzyme, is not sensitive to regulation by ATP at optimal pH 6.5 (Ranieri-Raggi et al. 1995).
The two Zn ions in the AMP deaminase metallocenter operate together as a catalytic unit, but play different roles in the catalytic mechanism, one of them (Zn1) acting to polarize the nucleophile water molecule, while the other (Zn2), possibly in co-operation with Zn1, could act transiently as the receptor for an activating substrate molecule (Fig. 3). The putative Zn2-binding site might represent the regulatory site at which the competition between activatory (ADP) and inhibitory (ATP) adenine nucleotides could take place. This peculiar kinetic property of rabbit skeletal muscle AMPD is likely to be due to the operation of a regulatory anion-binding site consisting of a cluster of positive charges localized between residues 72 and 80 (RKKRFQGRK, Fig. 3) (Martini et al. 2001).
The X-ray crystal structures of Arabidopsis t. and rabbit skeletal muscle AMPD have been recently resolved (Han et al. 2006; Bazin et al. 2003): the catalytic zinc is coordinated by three histidine and one aspartic acid residues (His-391, His-393, His-659, Asp-736 in Arabidopsis and His-303, His-305, His-572, Asp-649 in the rabbit) and these four aa residues align to those (His-15, His-17, His-214, Asp-295) that bind zinc in yeast adenosine deaminase (Wilson et al. 1991). The alignment with the AMPD protein sequences available at the NCBI website (http://www.ncbi.nlm.nih.gov/) shows that those aa residues are conserved in all eukaryotic species (data not shown). However, it should be pointed out that in both cases the AMPD crystals were obtained using an N-truncated protein, missing the aa residues that could coordinate the Zn2 ion in a dinuclear zinc site such that hypothesized for native rabbit skeletal muscle AMPD. It is likely therefore that the N-truncated protein could only bind the Zn1 ion with a coordination that differs from what has been described for Zn1 in the dinuclear zinc center (Fig. 3).
It has been shown that a histidine-proline-rich glycoprotein (HPRG) is associated strongly with purified rabbit skeletal muscle AMPD (Ranieri-Raggi et al. 1997). An anti-HPRG antibody selectively marked the type IIB fibers that contain the highest level of AMPD isoform M (Sabbatini et al. 1999). Furthermore, the immunological reaction of the anti-HPRG antibody and of an antibody specific to AMPD (isoform M) in human skeletal muscle biopsies from patients with AMPD deficiency clearly indicated a correlation between the muscle content of the HPRG-like protein and the level of AMPD activity (Sabbatini et al. 2006).
HPRG is a single chain glycoprotein that is present at a relatively high concentration in the plasma of vertebrates (100–150 µg/mL in humans) (Morgan 1978; Corrigan et al. 1990). HPRG is produced in the liver, but it has also been reported to be synthesized by monocytes, macrophages and megakaryocytes (Wakabayashi 2013). However, no HPRG mRNA was found in immune tissues by RT-PCR analysis, indicating that the HPRG present in immune cells is acquired from plasma (Hulett and Parish 2000). Sabbatini et al. (2011) reported that skeletal muscle cells do not transcribe HPRG but internalize the protein from serum. The percentage of internalized HPRG was reduced in the presence of heparin, indicating that heparin may compete with the cell surface GAGs in binding HPRG. These results are in accordance with other studies showing that human HPRG is actively internalized by the human T cell line MT4 and that the interaction of the protein with the cell membrane is mediated by GAGs (Olsen et al. 1996). Recent experiments with radiolabeled HPRG showed that the plasma half-life of HPRG was less than 15 min and that HPRG was internalized quickly in healthy tissues, including muscle, and in tumors (Tugues et al. 2014).
Plasma HPRG can bind divalent transition metal cations such as zinc, copper, mercury, cadmium and nickel. The interaction between HPRG and Zn2+ is abolished when histidine residues are chemically removed from HPRG, supporting the model that histidine residues are essential for Zn2+ binding (Morgan 1981).
While the physiological relevance of the interaction of plasma HPRG with Zn remains unclear, evidence has been given of a Zn-specific binding to HPRG isolated from the rabbit skeletal muscle AMPD complex (Ranieri-Raggi et al. 2014). The EXAFS analysis of a 2:1 Zn–HPRG complex showed that HPRG might bind two metal ions at about 3.7 Å distance, consistent with the presence of a dinuclear first transition-row metal site bridged by side-chain groups like carboxylates or cysteine thiolate sulfur (Mangani et al. 2003). Based on the EXAFS data six amino acid residues (i.e., Asp-252, His-253, His-255, His-257, His-260 and Cys-264) were proposed to chelate zinc and contribute to the formation of a specific dinuclear binding site in the 252–264 residue region of rabbit HPRG (Ronca and Raggi 2015). A second hypothetical dinuclear zinc-binding site model could involve both Cys-264 and Cys-294, located in the 252–310 residue region within the PRR1 region. Two possible four-coordinate zinc sites are therefore envisioned, where one cysteine sulfur and three among aspartate or glutamate carboxylate groups and histidine imidazoles might chelate the metal (Ronca and Raggi, 2015).
By coupling the finding that rabbit muscle HPRG as well as the catalytic subunit of AMPD is able to bind zinc in a dinuclear metal binding site with the previous suggestion of the presence in the whole AMP deaminase of additional zinc not required for activity (Zielke and Suelter 1971) and by considering the absence of significant differences in the kinetics of AMPD with different HPRG content (Mangani et al. 2003), the addition of HPRG into the family of metallochaperones has been envisaged, suggesting that HPRG may enhance the stability of AMPD in vivo through insertion of zinc or by modulating the intracellular zinc availability (Mangani et al. 2003). This view is strengthened by the results of an investigation of the effects of the isolation by zinc-affinity chromatography of the HPRG component of rabbit skeletal muscle AMPD (Ranieri-Raggi et al. 2003). When the whole enzyme was loaded on a Zn2+_charged affinity column under denaturing and reducing conditions, only the HPRG component was specifically retained on the column, while the catalytic subunit emerged in the void volume and precipitated immediately in the flow-through. The protein precipitation has been ascribed to a disruption of the association between the two components of the enzyme due to the selective binding of HPRG to the resin. HPRG was only eluted with an EDTA-containing buffer that strips Zn2+ from the gel (Ranieri-Raggi et al. 2003).
A heterotetramer model for the AMPD/HPRG complex (i.e., a dimer of approximately 155 kDa heterodimers) has been envisaged (Mangani et al. 2003).
5 Evidence of an in vitro regulatory action of TnT on AMPD
Since the rabbit skeletal muscle apparatus is generally composed of white muscle, rabbit skeletal muscle AMPD preparations show the considerably higher specific activity of the isoform specific to white muscle (isoform M) at physiological concentration of AMP (0.1 mM) which shows a peculiar inhibition by ATP, especially at pH values higher than neutrality, with ADP being the most efficient metabolite in counteracting that inhibition (Ronca-Testoni et al. 1970). Upon storage at 4 °C, rabbit skeletal muscle AMP deaminase undergoes a partial proteolysis that increases the activity of the enzyme and modifies its regulation by adenine nucleotides. Cleavage of purified AMPD during storage is produced by a calpain-like proteinase that converts the native 85-kDa enzyme subunit into a 75-kDa core that is resistant to further degradation (Ranieri-Raggi and Raggi 1980; Martini et al. 2004). A very similar pattern of digestion and enzymatic activation is obtained by limited proteolysis by trypsin that removes the 95-residue N-terminal domain of AMPD (Ranieri-Raggi and Raggi 1979; Ronca et al. 1994). When the enzyme is incubated with trypsin at a ratio of 1/100 trypsin/AMPD, a rapid activation of the enzyme at low substrate concentration is observed. The increase in activity of AMPD by trypsin is due to the transition of the enzyme to a form that follows hyperbolic substrate–velocity curve, whereas the native enzyme shows a sigmoidal one (Fig. 4a).

Regulation of rabbit fast skeletal muscle AMPD. Effects of limited proteolysis on the enzyme substrate versus velocity curve (a) and on the modulation of the enzyme by ADP (b), ATP (c) and GTP (d). The reaction mixture contained 50 mM imidazole–HCl (pH 6.5), 3 mM KCl and the reported AMP concentrations (a) or 0.1 mM AMP (b–d). (filled circle) Native enzyme; (unfilled circle) trypsin-treated enzyme
In the proteolysed enzyme, the effects of ADP and ATP on AMPD are also markedly modified: while ADP activates the native enzyme, reverting sigmoidal to hyperbolic kinetics (not shown), the trypsin-treated enzyme shows an almost complete loss of sensitivity toward this nucleotide (Fig. 4b). The trypsin-treated enzyme is no longer inhibited by ATP, whereas the inhibitory effect of GTP appears to be unmodified after proteolysis (Fig. 4c, d).
Upon the observations that AMPD is a contaminant of actomyosin preparations and that it can be dissociated from actomyosin by inorganic phosphate (Currie and Webster 1962), we studied the interaction in vitro of AMPD with several proteins of the contractile apparatus and their influence on both the activity and regulation of the enzyme.
We have observed that the addition of small amounts of a crude preparation of actomyosin (Currie and Webster 1962) from rabbit white skeletal muscle restores in AMPD the allosteric properties that are removed by limited proteolysis with trypsin. When crude actomyosin, in a ratio of about 10:1 actomyosin/AMPD (w/w), was added to the enzyme assay in the presence of low K+ concentration, the proteolysed enzyme showed a reduction of the activity to the level shown by the native enzyme in the same conditions (Fig. 5a). Thus, the hyperbolic kinetic curve of the trypsin-treated AMPD reverted to the sigmoidal one shown by the native enzyme (Fig. 5c). No significant effect by actomyosin was observed on the activity of the native enzyme (Fig. 5a).

Effects of crude actomyosin and TnT on the activity of trypsinized rabbit fast skeletal muscle AMPD. a AMPD activity at 3 mM KCl as a function of crude actomyosin concentration; (filled circle) 5 μg/ml native enzyme; (unfilled circle) 5 μg/ml trypsin-treated enzyme. b Activity of trypsin-treated AMPD (2.5 nM) at 3 mM (unfilled circle) or 60 mM (filled circle) KCl as a function of TnT concentration. a, b The reaction mixtures contained 50 mM imidazole–HCl (pH 6.5) and 0.1 mM AMP. c Substrate versus velocity curves of 5 μg/ml trypsin-treated AMPD alone (filled circle) or in the presence of 20 μg/ml (filled square) or 100 μg/ml (filled triangle) crude actomyosin. d Substrate versus velocity curves of 2.5 nM trypsin-treated AMPD alone (filled circle) or in the presence of 20 nM (unfilled circle) or 50 nM (unfilled triangle) TnT. c, d The reaction mixtures contained 50 mM imidazole–HCl (pH 6.5), 3 mM KCl and the reported AMP concentrations
The effect of individual proteins of the actomyosin complex on the activity of native and proteolysed AMPD was also tested. While the activities of both native and proteolysed AMPD were not affected either by the presence of heavy or light myosin chains, tropomyosin, TnC or TnI (not shown), the addition of rabbit skeletal muscle TnT (Ranieri-Raggi et al. 1985) induced a marked reduction of the activity of trypsin-treated AMP deaminase at low substrate concentration which decreased with increasing TnT concentration until a ratio of about 10 mol TnT per mol enzyme was attained (Fig. 5b). This inhibition caused the hyperbolic substrate–velocity curve shown by the proteolysed enzyme to revert to the sigmoid one shown by the native enzyme (Fig. 5d) (Ranieri-Raggi et al. 1985).
To test whether the whole protein or a specific region of TnT is required for the modulation of the AMP deaminase activity, TnT was digested with trypsin and the peptides of TnT were separated by chromatography on Sephadex G-50 following the method of Moir et al. (1977). The activity of trypsinized AMPD was clearly inhibited only by the addition of the first peak of the eluted fractions (Fig. 6a) whose SDS/PAGE and N-terminal sequence analyses showed the presence of a single component of approximately 5 kDa corresponding to the 50-aa N-terminal TnT peptide (see Fig. 1). The N-terminal peptide inhibited the activity of proteolysed AMPD, reproducing the effect exerted by the addition of intact TnT (Fig. 6b). The addition of the N-terminal peptide of TnT restores the allosteric properties of the native enzyme that were removed by the limited proteolysis, i.e., activation by ADP (Fig. 6c) and inhibition by ATP (Fig. 6d). The N-terminal peptide, as well as whole TnT, exerted no effect on either the kinetic or the regulatory properties of native AMPD (not shown). It should be also pointed out that in the presence of activating K+ concentration (60 mM KCl), the activity of the trypsinized enzyme is not affected by the addition of whole TnT or TnT N-terminal peptide (Fig. 6b).

Effect of TnT fragments on the activity of trypsinized AMPD. A tryptic digest of TnT was applied to a Sephadex G-50 column in 0.01 M HCl and 5.6 ml fractions were collected. a The solid line (-) is the A215 of each sample and (unfilled circle) is the activity of trypsin-treated AMPD in the presence of 50 mM imidazole–HCl (pH 6.5), 0.1 mM AMP, 3 mM KCl and 100 μl of various fractions. The horizontal bar indicates the elution position of the TnT N-terminal peptide. b Activity of trypsinized AMPD, 2.5 nM, as a function of the concentration of the N-terminal peptide of TnT (0–100 nM). The reaction mixture contained 50 mM imidazole–HCl (pH 6.5), 0.1 mM AMP and 3 mM (unfilled circle) or 60 mM (filled circle) KCl. The activity of trypsinized AMPD, 2.5 nM, as a function of ADP (c) or ATP (d) concentration. The reaction mixture contained 50 mM imidazole–HCl (pH 6.5), 3 mM KCl and 0.1 mM AMP in the absence (filled circle) or in the presence (unfilled circle) of 75 nM TnT N-terminal peptide
It is known that TnT is isolated from the troponin complex of striated muscle as a phosphorylated form, mainly at serine-1 (Perry 1998; Moir et al. 1977). Interestingly, upon treatment with alkaline phosphatase, the N-terminal peptide of troponin T loses its capability to inhibit trypsinized AMPD and no longer influences the modulation of the allosteric properties of AMPD by ADP and ATP (Ranieri-Raggi et al. 1985).
6 Restrictive proteolysis by calpain at the N-terminal region of fast TnT and AMPD removes a putative regulatory zinc-binding site: contribution of the enzyme to the regulation of muscle contraction exerted by TnT
An intriguing correlation between AMPD and TnT is that some isoforms of both proteins undergo restrictive proteolysis by calpain protease.
Rabbit AMD1 is cleaved in vitro by a protease that releases a 97 aa N-terminal fragment and shows a specificity identical to that reported for ubiquitous calpains (Ranieri-Raggi and Raggi 1980; Martini et al. 2004). A similar effect of storage has been reported on human AMPD1 (skeletal muscle) and AMPD3 (cardiac muscle) recombinant isoenzymes that are proteolysed to truncated polypeptides ∆I86 and ∆H98, and ∆L88 and ∆M90, respectively, all cleavage sites having Leu at P2, suggesting that a calpain-mediated proteolytic cleavage plays a significant role in the regulation of AMPD activity in striated muscle (Mahnke-Zizelman et al. 1998). The rabbit-cleaved enzyme is more active at low substrate concentration and no longer inhibited by ATP, similarly to the effect of the removal of the 95 aa N-terminal domain by trypsin (Ranieri-Raggi and Raggi 1979). Although the function and the mechanisms of calpain proteolysis of AMPD are unknown, it has been hypothesized that this phenomenon could be responsible for the large ammonia accumulation during episodes of strong tetanic contraction of skeletal muscle or during rigor mortis (Bendall and Davey 1957). Interestingly, the rabbit AMPD1 N-terminal domain contains a calpastatin-like sequence that might protect against an unrestrained activation of the enzyme that otherwise would lead to the depletion of adenine nucleotide stores (Martini et al. 2004).
An in vivo calpain-induced degradation was observed with cardiac and skeletal muscle TnT. Restricted proteolysis of cardiac TnT by calpain occurs during physiological and pathological adaptation of cardiac muscle, such as ischemia–reperfusion and pressure overload (Zhang et al. 2006; Feng et al. 2008). Upon the selective removal of the 71 aa N-terminal variable region by m-calpain, the cardiac TnT-binding affinities for Tm, TnC and TnI were altered and a lower myosin ATPase activity and myofibril force generation were observed. More recently, a non-canonical role for fast-twitching skeletal muscle TnT was reported by Zhang et al. (2016). TnT is fragmented in aging mice and both the full-length TnT and the C-terminal domain of TnT can shuttle to the nucleus. Here, the expression of the voltage sensor Ca2+ channel a1 subunit, partially responsible for the loss of muscle strength during aging, is increased in the presence of TnT but is decreased by its C-terminal fragment.
Although it is yet to be established whether the proteolysis of AMPD and TnT by calpain could have either a regulatory role or a degradative one, data from our laboratory have reported that rabbit fast skeletal TnT or its phosphorylated 50-residue N-terminal peptide restores in rabbit skeletal muscle AMPD the allosteric properties removed by limited proteolysis by trypsin in vitro as well as by a calpain-like proteinase in vivo (Ranieri-Raggi et al. 1985; Martini et al. 2004). Based on these observations of the effects of the interaction between AMPD and TnT in vitro on the enzyme kinetics and modulation, we advance the hypothesis that the unrestrained AMPD activity that follows the proteolytic cleavage of the enzyme, occurring in strenuously exercised muscle (Fig. 7a), could be counteracted by the binding of the 50-residue N-terminal domain of TnT to the catalytic zinc metallocenter of AMPD (Fig. 7b). Therefore, TnT might mimic the regulatory action exerted in AMPD by the cleavable 95-residue N-terminal domain that holds the native enzyme in a less active conformation due to the formation of a dinuclear zinc active center (Fig. 3), causing the enzyme to exhibit both the positive homotropic behavior (i.e., activation by the substrate AMP) and the allosteric regulation by nucleoside di- and triphosphates. This hypothesis is strengthened by the observation that the N-terminal region of TnT in some isoforms reveals the presence of a zinc-binding segment. In particular, in rabbit fast TnT (Fig. 1), the sequence of residues 29–50 (29HEPAPEVHVPEEVHEDALEDMR50) contains two putative zinc-binding sites (HEXXXE) that are compatible with the HEXXXH sequence in the N-terminal region of rabbit AMPD that has been shown to bind zinc (Mangani et al. 2007). In this context also, it should be taken into consideration the effects that the TnT cleavage by calpain could exert on the modulation of AMPD as well as on myofibril force generation.

Involvement of the N-terminal Zn-binding regions of rabbit fast AMPD and TnT in the modulation of the enzyme activity. Models of the AMPD catalytic subunit (above) and schema of the components of the muscle thin filament (below) with the N-terminal domain of the TnT protein depicted according to Amarasinghe and Jin (2015) (a). The scissors indicate the sites of the in vivo proteolytic activity of calpains on AMPD and TnT that releases the Zn-binding N-terminal domains of both proteins. The amino acid residues involved in the binding of the two Zn ions in the metallocenter of native AMPD are shown in Fig. 3; those presumably involved in the binding of Zn to TnT belong to the region identified in Fig. 1. Native AMPD, in pink, is an allosteric regulated enzyme that exhibits sigmoid kinetics in the presence of ATP and is activated by AMP and ADP. Upon proteolysis by calpain that might occur during strenuous exercise, cleaved AMPD, in red, shows an unrestrained activity, following hyperbolic kinetics, and it is no longer modulated by adenine nucleotides. The coordination of the zinc ion in proteolyzed AMPD is based on the model proposed for the catalytic site of adenosine deaminase (Wilson et al. 1991). TnT N-terminal domain inhibits the activity of proteolyzed AMPD, restoring the sigmoid kinetics and the allosteric properties of the native enzyme, i.e., activation by ADP and inhibition by ATP (see Fig. 6) (b) (color figure online)
A repeating transition metal-binding histidine-rich sequence, designated TX and consisting of four- or seven-repeated segment HE/AEAH has been identified in the hypervariable N-terminal region of TnT isoforms in the breast but not leg muscles of all Galliformes, while it was absent in all other species of birds investigated (Jin and Smillie 1994; Jin and Root 2000). It was observed that the affinity of TX for zinc was adequate to affect free metal levels in the muscle cell and that a link may exist between the presence of the TX sequence, the high muscle content of fast glycolytic fiber type in pectoralis muscle and the explosive but short-lived flight pattern of these birds. Structural changes induced by Zn2+ binding to the TX segment of chicken breast TnT altered the binding affinity of TnT to Tm (Ogut and Jin 1998). In addition, the His repeat domain may be sensitive to alterations in cellular pH that drops from approximately 7.1 to 6.5 during anaerobic work, since histidine-imidazole groups are half-protonated at those pH values and might therefore be involved in reversible protonation–deprotonation events that could control the conformation of the N-terminal domain of TnT (Bucher et al. 1999). The binding of fast TnT to Tm has actually been shown to be more stable at lower pH (Ogut and Jin 1998). However, the metal ion-induced changes would only occur in that class of TnT isoforms and its biological effect in the avian breast muscle remains to be established.
In higher vertebrates, three homologous TnT genes encode for a large number of TnT isoforms that are differentially expressed in fast skeletal, slow skeletal and cardiac muscle fibers. These isoforms differ mainly in the sequence, the length and the charge of the N-terminal region while the central and C-terminal regions are highly conserved. Although the relationship between the structural variations of the N-terminal region and its functional role in specific type of muscle cells is yet to be determined, it has been reported that the variable N-terminal region does not bind any known myofilament proteins and it has been proposed that it modulates the binding affinity of the TnT core protein to TnC, TnI and Tm and the Ca2+ sensitivity during muscle contraction (Amarasinghe and Jin 2015). Interestingly, only some of the fast skeletal TnT variants, generated by alternative splicing, contain one or two putative Zn-binding sites within the N-terminal region. The ability of the TnT variable N-terminal region to interact with zinc and with other proteins such as AMPD could be at the base of a wide range of physiological responses of different muscle fibers, allowing a rapid and fine-tuned adaptation to the changes in contractile demands.
7 Relationship between the AMPD activity and muscle specialization: interaction between AMPD and myofibrils
Multiple isoforms of AMPD have been purified and characterized from rat, rabbit and human tissues. The isoenzymes are encoded by three mammalian genes, i.e., ampd1, that is highly expressed in skeletal muscle, and ampd2 and ampd3, the smooth and cardiac muscle isoforms, respectively, although both are also expressed in various other tissues (Sabina et al. 1990). In addition to this diversification in the isoenzyme expression in different muscle and fiber types, among AMPD1, AMPD2 and AMPD3 isoenzymes the N-terminal domains are highly divergent, while the C-terminal domains are highly conserved. Among the AMPD1 homolog sequences, the rabbit and the human enzymes, but not the rat sequence, contain a putative zinc-binding site in the N-terminal region. The sequence differences confer different catalytic and regulatory properties to the different isoenzymes suggesting that specialization might have occurred in the different muscle types based on the fiber composition as well as on the animal activity. This view is supported by the following observations: in the pigeon the muscles of the wing, that have a higher capacity to derive energy from glycolytic reactions also have about four times AMPD and two times AK in comparison with the enzyme activities present in leg muscles (Raggi et al. 1969). In the bat, the muscles of the wing and the pectoralis contain an AMPD activity four to five times higher than that of the diaphragm and hind limbs. However, the pectoralis and diaphragm have a higher oxidative metabolism compared to that of the wing and hind limb muscles (Raggi et al. 1974). These observations indicate that the AMPD activity could be associated with a specific type of muscle specialization and mechanics rather than with the metabolic characteristics of the muscle.
Based on the early reports that AMPD is a persistent contaminant of actomyosin preparations (Currie and Webster 1962), several investigators have established that the AMPD activity is associated with various elements of contractile fibers. AMPD has been found to bind native myosin and purified myosin S2 fragments (Ashby and Frieden 1977; Ashby et al. 1979; Koretz and Frieden 1980). The reversible interaction between AMPD and myofibrils is promoted in vivo by intense muscle contraction and results in an increase in AMPD activity, suggesting that the enzyme activation may help to maintain the adenylate charge and preserve cell viability under stressful conditions (Shiraki et al. 1981; Rundell et al. 1992a, b, 1993). However, it has also been reported that AMPD binds titin with a 12-nm periodicity, although the significance of this interaction remains unclear (Koretz et al. 1993). More recently, Mahnke-Zizelman and Sabina (2001) reported that the binding capacity of AMPD isoforms to actomyosin resides in the C-terminal region of the enzyme, but N-truncated forms of AMPD1 exibits a significantly lower capacity to bind actomyosin, raising the question whether the AMPD interaction data previously described were affected by the degree of N-terminal proteolysis in the preparations.
Somewhat different observations were obtained when the localization of AMPD in skeletal muscle was identified by immunostaining methods with both isolated myofibrils and muscle fibers grown in culture. In unstretched chicken and human muscle myofibrils, AMPD is bound to the lateral ends of the A-band extending into the I-band (Ashby et al. 1979). In addition, AMPD staining appears as a thin line in the center of the A-band, in correspondence to the M-line (van Kuppevelt et al. 1994) where CK and AK reside (Hu et al. 2015). In rabbit fast-twitch muscles, the AMPD immunostaining was located in the A-band in a region significantly beyond the end of the myosin filament. It has been suggested that AMPD may be attached to myofibrils through titin molecules that extend from the tip of the thick filament to the Z-line and that this immobilization of the enzyme in the sarcomere could improve its metabolic efficiency (Cooper and Trinick 1984). It should be pointed out, however, that no clear influence on AMPD activity has been found as a result of the interaction of the enzyme with either titin or myosin. In contrast, a functional role for skeletal muscle AMPD can be proposed that is based on the results reported in this review (see Sect. 5) on the relationship between the enzyme and fast TnT.
8 Conclusions
During muscle contraction, AMPD acts to displace the AK equilibrium to generate ATP and AMP from two ADP molecules; the ATP produced is then readily available to myosin ATPase.
The N-terminus of AMPD shares with calpastatin a regulatory domain that during normoxic contractions might impede access of calpain to its site of action, thereby exerting a protective role against the protease-induced fragmentation of AMPD. However, in skeletal muscle subjected to strong tetanic contraction a calpain-induced activation of the enzyme occurs that is responsible for the large ammonia accumulation observed during sustained muscle activity.
The unrestrained AMPD activity that follows the proteolytic cleavage of the enzyme during strenuous exercise could be counteracted by the binding of AMPD to the phosphorylated N-terminal region of TnT that could restore the less active conformation of the native enzyme, possibly through the interaction of the catalytic Zn ion present at the C-terminus of the enzyme with the putative metal-binding site found at the N-terminal regulatory region of TnT. In this view, TnT mimics the regulatory action exerted in AMPD by the 95-residue N-terminal domain that holds the enzyme in a less active conformation due to the presence of the dinuclear zinc center, causing the enzyme to exhibit both the positive homotropic behavior (i.e., activation by the substrate AMP) and the modulation by adenosine di- and triphosphates (i.e., activation by ADP and inhibition by ATP).
We conclude that all together the observations reported in this review on the interactions between AMPD and TnT strongly suggest that the two proteins might exert a mutual contribution to the regulation of muscle contraction.
References
Amarasinghe C, Jin JP (2015) N-Terminal hypervariable region of muscle type isoforms of troponin T differentially modulates the affinity of tropomyosin-binding site 1. Biochemistry 54:3822–3830
Amphlett GW, Vanaman TC, Perry SV (1976) Effect of the troponin C-like protein from bovine brain (brain modulator protein) on the Mg2+-stimulated ATPase of skeletal muscle actomyosin. FEBS Lett 72:163–168
Ashby B, Frieden C (1977) Interaction of AMP-aminohydrolase with myosin and its subfragments. J Biol Chem 252:1869–1872
Ashby B, Frieden C, Bischoff R (1979) Immunofluorescent and histochemical localization of AMP deaminase in skeletal muscle. J Cell Biol 81:361–373
Atkinson DE (1968) The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 7:4030–4034
Barsacchi R, Ranieri-Raggi M, Bergamini C, Raggi A (1979) Adenylate metabolism in the heart. Regulatory properties of rabbit cardiac adenylate deaminase. Biochem J 182:361–366
Bazin RJ, MacDonald GA, Phillips C (2003) Adenosine monophosphate deaminase crystal structure. UK Patent GB2373504
Bendall JR, Davey CL (1957) Ammonia liberation during rigor mortis and its relation to changes in the adenine and inosine nucleotides of rabbit muscle. Biochim Biophys Acta 26:93–103
Biesiadecki BJ, Chong SM, Nosek TM, Jin JP (2007) Troponin T core structure and the regulatory NH2-terminal variable region. Biochemistry 46:1368–1379
Bockman EL, McKenzie JE (1979) Adenosine production in skeletal muscles of different fibre types. In: Baer HP, Drummond GI (eds) Physiological and regulatory functions of adenosine and adenine nucleotides. Raven Press, New York, pp 145–153
Breitbart RE, Nguyen HT, Medford RM, Destree AT, Mahdavi V, Nadal-Ginard B (1985) Intricate combinatorial patterns of exon splicing generate multiple regulated troponin T isoforms from a single gene. Cell 41:67–82
Bucher EA, Dhoot GK, Emerson MM, Ober M, Emerson CP Jr (1999) Structure and evolution of the alternatively spliced fast troponin T isoform gene. J Biol Chem 274:17661–17670
Conway EJ, Cooke R (1939) The deaminases of adenosine and adenylic acid in blood and tissues. Biochem J 33:479–492
Cooper J, Trinick J (1984) Binding and location of AMP deaminase in rabbit psoas muscle myofibrils. J Mol Biol 177:137–152
Corrigan JJ, Jeter MA, Bruck D, Feinberg WM (1990) Histidine-rich glycoprotein levels in children: the effect of age. Thromb Res 59:681–686
Currie RD, Webster HL (1962) Preparation of 5′-adenylic acid deaminase based on phosphate-induced dissociation of rat actomyosin-deaminase complexes. Biochim Biophys Acta 64:30–40
Davey CL (1961) The amination of inosine monophosphate in skeletal muscle. Arch Biochem Biophys 95:296–304
Dobroval’skiĭ AB, Gusev NB, Martinov AV, Severin SE (1976) Search for a protein kinase specific for troponin T. Biokhimiya 41:1291–1296
Feng HZ, Biesiadecki BJ, Yu ZB, Hossain MM, Jin JP (2008) Restricted N-terminal truncation of cardiac troponin T: a novel mechanism for functional adaptation to energetic crisis. J Physiol 586:3537–3550
Gomes AV, Potter JD, Szczesna-Cordary D (2002) The role of troponins in muscle contraction. IUBMB Life 54:323–333
Gomes AV, Venkatraman G, Davis JP, Tikunova SB, Engel P, Solaro RJ, Potter JD (2004) Cardiac troponin T isoforms affect the Ca(2+) sensitivity of force development in the presence of slow skeletal troponin I: insights into the role of troponin T isoforms in the fetal heart. J Biol Chem 279:49579–49587
Gordon AM, Homsher E, Regnier M (2000) Regulation of contraction in striated muscle. Physiol Rev 80:853–924
Han BW, Bingman CA, Mahnke DK, Bannen RM, Bednarek SY, Sabina RL, Phillips GN Jr (2006) Membrane association, mechanism of action, and structure of Arabidopsis embryonic factor 1 (FAC1). J Biol Chem 281:14939–14947
Hartshorne DJ, Perry SV (1962) A chromatographic and electrophoretic study of sarcoplasm from adult- and foetal-rabbit muscles. Biochem J 85:171–177
Heeley DH, Golosinska K, Smillie LB (1987) The effects of troponin T fragments T1 and T2 on the binding of non polymerizable tropomyosin to F-actin in the presence and absence of troponin I and troponin C. J Biol Chem 262:9971–9978
Hu LY, Ackermann MA, Kontrogianni-Konstantopoulos A (2015) The sarcomeric M-region: a molecular command center for diverse cellular processes. Biomed Res Int 2015:714197
Hulett MD, Parish CR (2000) Murine histidine-rich glycoprotein: cloning, characterization and cellular origin. Immunol Cell Biol 78:280–287
Jackson P, Amphlett GW, Perry SV (1975) The primary structure of troponin T and the interaction with tropomyosin. Biochem J 151:85–97
Jin JP (2016) Evolution, regulation, and function of N-terminal variable region of troponin T: modulation of muscle contractility and beyond. Int Rev Cell Mol Biol 321:1–28
Jin JP, Chong SM (2010) Localization of the two tropomyosin-binding sites of troponinT. Arch Biochem Biophys 500:144–150
Jin JP, Root DD (2000) Modulation of troponin T molecular conformation and flexibility by metal ion binding to the NH2-terminal variable region. Biochemistry 39:11702–11713
Jin JP, Smillie LB (1994) An unusual metal-binding cluster found exclusively in the avian breast muscle troponin T of Galliformes and Craciformes. FEBS Lett 341:135–140
Jin JP, Huang QQ, Yeh HI, Lin JJ (1992) Complete nucleotide sequence and structural organization of rat cardiac troponin T gene. A single gene generates embryonic and adult isoforms via developmentally regulated alternative splicing. J Mol Biol 227:1269–1276
Kendrick-Jones J, Perry SV (1967) The enzymes of adenine nucleotide metabolism in developing skeletal muscle. Biochem J 103:207–214
Koretz JF, Frieden C (1980) Adenylate deaminase binding to synthetic thick filaments of myosin. Proc Natl Acad Sci 77:7186–7188
Koretz JF, Irving TC, Wang K (1993) Filamentous aggregates of native titin and binding of C-protein and AMP-deaminase. Arch Biochem Biophys 304:305–309
Lowenstein JM (1972) Ammonia production in muscle and other tissues: the purine nucleotide cycle. Physiol Rev 52:382–414
Lowenstein JM, Tornheim K (1971) Ammonia production in muscle: the purine nucleotide cycle. Science 171:397–400
Mahnke-Zizelman DK, Sabina RL (2001) Localization of N-terminal sequences in human AMP deaminase isoforms that influence contractile protein binding. Biochem Biophys Res Commun 285:489–495
Mahnke-Zizelman DK, Tullson PC, Sabina RL (1998) Novel aspects of tetramer assembly and N-terminal domain structure and function are revealed by recombinant expression of human AMP deaminase isoforms. J Biol Chem 273:35118–35125
Malnic B, Farah CS, Reinach FC (1998) Regulatory properties of the NH2- and COOH-terminal domains of troponin T. ATPase activation and binding to troponin I and troponin C. J Biol Chem 273:10594–10601
Mangani S, Meyer-Klaucke W, Moir AJG, Ranieri-Raggi M, Martini D, Raggi A (2003) Characterization of the Zn binding site of the HPRG protein associated with rabbit skeletal muscle AMP deaminase. J Biol Chem 278:3176–3184
Mangani S, Benvenuti M, Moir AJG, Ranieri-Raggi M, Martini D, Sabbatini ARM, Raggi A (2007) Characterization of the metallocenter of rabbit skeletal muscle AMP deaminase. Evidence for a dinuclear zinc site. Biochim Biophys Acta 1774:312–322
Marquetant R, Desai NM, Sabina RL, Holmes EW (1987) Evidence for sequential expression of multiple AMP deaminase isoforms during skeletal muscle development. Proc Natl Acad Sci 84:2345–2349
Martini D, Ranieri-Raggi M, Sabbatini ARM, Raggi A (2001) Regulation of skeletal muscle AMP deaminase: lysine residues are critical for the pH-dependent positive homotropic cooperativity behaviour of the rabbit enzyme. Biochim Biophys Acta 1544:123–132
Martini D, Montali U, Ranieri-Raggi M, Sabbatini ARM, Thorpe SJ, Moir AJ, Raggi A (2004) A calpain-like proteolytic activity produces the limited cleavage at the N-terminal regulatory domain of rabbit skeletal muscle AMP deaminase: evidence of a protective molecular mechanism. Biochim Biophys Acta 1702:191–198
Martini D, Ranieri-Raggi M, Sabbatini ARM, Moir AJG, Polizzi E, Mangani S, Raggi A (2007) Characterization of the metallocenter of rabbit skeletal muscle AMP deaminase. A new model for substrate interactions at a dinuclear cocatalytic Zn site. Biochim Biophys Acta 1774:1508–1518
Medford RM, Nguyen HT, Destree AT, Summers E, Nadal-Ginard B (1984) A novel mechanism of alternative RNA splicing for the developmentally regulated generation of troponin T isoforms from a single gene. Cell 38:409–421
Merkler DJ, Schramm VL (1993) Catalytic mechanism of yeast adenosine 5′-monophosphate deaminase. Zinc content, substrate specificity, pH studies and solvent isotope effects. Biochemistry 32:5792–5799
Moir AJ, Cole HA, Perry SV (1977) The phosphorylation sites of troponin T from white skeletal muscle and the effects of interaction with troponin C on their phosphorylation by phosphorylase kinase. Biochem J 161:371–382
Morgan WT (1978) Human serum histidine-rich glycoprotein. I. Interactions with heme, metal ions and organic ligands. Biochim Biophys Acta 535:319–333
Morgan WT (1981) Interactions of the histidine-rich glycoprotein of serum with metals. Biochemistry 20:1054–1061
Newton AA, Perry SV (1960) The incorporation of 15N into adenine nucleotides and their formation from inosine monophosphate by skeletal muscle preparations. Biochem J 74:127–136
Ogut O, Jin JP (1998) Developmentally regulated, alternative RNA splicing-generated pectoral muscle-specific troponin T isoforms and role of the NH2-terminal hypervariable region in the tolerance to acidosis. J Biol Chem 273:27858–27866
Ohtsuki I (1979) Molecular arrangement of troponin-T in the thin filament. J Biochem 86:491–497
Ohtsuki I, Shiraishi F, Suenaga N, Miyata T, Tanokura M (1984) A 26 K fragment of troponin T from rabbit skeletal muscle. J Biochem 95:1337–1342
Olsen HM, Parish CR, Altin JG (1996) Histidine-rich glycoprotein binding to T-cell lines and its effect on T-cell substratum adhesion is strongly potentiated by zinc. Immunology 88:198–206
Pan BS, Gordon AM, Potter JD (1991) Deletion of the first 45 NH2-terminal residues of rabbit skeletal troponin T strengthens binding of troponin to immobilized tropomyosin. J Biol Chem 266:12432–12438
Parnas JK (1929) Über die Ammoniakbildung im Muskel und ihren Zusammenhang mit Funktion und Zustandsänderung. Der Zusammenhang der Ammoniakbildung mit der Umwandlung des Adeninnucleotids zu Inosinsäure. Biochem Z 206:16–38
Parnas JK, Lewinski W (1935) Über den Zusammenhang zwischen Ammoniakbildung und Muskeltätigkeit unter aeroben Bedingungen. Biochem Z 276:398–407
Parnas JK, Lewinski W, Jaworska J, Umschweif B (1930) Über den Ammoniakgehalt und die Ammoniakbildung im Froschmuskel. Biochem Z 228:366–400
Pearlstone JR, Smillie LB (1977) The binding site of skeletal alpha-tropomyosin on troponin T. Can J Biochem 55:1032–1038
Pearlstone JR, Smillie LB (1978) Troponin T fragments: physical properties and binding to troponin C. Can J Biochem 56:521–527
Pearlstone JR, Smillie LB (1981) Identification of a second binding region on rabbit skeletal troponin-T for α-tropomyosin. FEBS Lett 128:119–122
Pearlstone JR, Smillie LB (1982) Binding of troponin-T fragments to several types of tropomyosin: sensitivity to Ca2+ in the presence of troponin C. J Biol Chem 257:10587–10592
Pearlstone JR, Carpenter MR, Smillie LB (1977) Primary structure of rabbit skeletal muscle troponin-T. Purification of cyanogen bromide fragments and the amino acid sequence of fragment CB2. J Biol Chem 252:971–977
Perry SV (1998) Troponin T: genetics, properties and function. J Muscle Res Cell Motil 19:575–602
Perry SV (1999) Troponin I: inhibitor or facilitator. Mol Cell Biochem 190:9–32
Perry SV (2001) Vertebrate tropomyosin: distribution, properties and function. J Muscle Res Cell Motil 22:5–49
Potter JD, Sheng Z, Pan BS, Zhao J (1995) A direct regulatory role for troponin T and a dual role for troponin C in the Ca2+ regulation of muscle contraction. J Biol Chem 270:2557–2562
Raggi A, Ranieri-Raggi M (1987) Regulatory properties of AMP deaminase isoenzymes from rabbit red muscle. Biochem J 242:875–879
Raggi A, Ronca-Testoni S, Ronca G (1969) Muscle AMP aminohydrolase. II. Distribution of AMP aminohydrolase, myokinase and creatine kinase activities in skeletal muscle. Biochim Biophys Acta 178:619–622
Raggi A, Ranieri M, Taponeco G, Ronca-Testoni S, Ronca G, Rossi CA (1970) Interaction of the rat muscle AMP aminohydrolase with chelating agents and metal ions. FEBS Lett 10:101–104
Raggi A, Bergamini C, Lunardini M (1974) Distribuzione dell’ AMP deaminasi nei muscoli scheletrici del pipistrello, Rinolophus ferrum-equinum. Boll Soc Ital Biol Sper 50:515–520
Raggi A, Bergamini C, Ronca G (1975) Isozymes of AMP deaminase in red and white skeletal muscles. FEBS Lett 58:19–23
Raggi A, Grand RJ, Moir AJ, Perry SV (1989) Structure-function relationships in cardiac troponin T. Biochim Biophys Acta 997:135–143
Ranieri-Raggi M, Raggi A (1979) Regulation of skeletal muscle AMP deaminase. Effects of limited proteolysis on the activity of the rabbit enzyme. FEBS Lett 102:59–63
Ranieri-Raggi M, Raggi A (1980) Effects of storage on activity and subunit structure of rabbit skeletal-muscle AMP deaminase. Biochem J 189:367–368
Ranieri-Raggi M, Moir AJ, Raggi A (1985) Interaction with troponin T from white skeletal muscle restores in white skeletal muscle AMP deaminase those allosteric properties removed by limited proteolysis. Biochim Biophys Acta 827:93–100
Ranieri-Raggi M, Ronca F, Sabbatini A, Raggi A (1995) Regulation of skeletal muscle AMP deaminase: involvement of histidine residues in the pH-dependent inhibition of the rabbit enzyme by ATP. Biochem J 309:845–852
Ranieri-Raggi M, Montali U, Ronca F, Sabbatini A, Brown PE, Moir AJG, Raggi A (1997) Association of purified skeletal-muscle AMP deaminase with a histidine-proline-rich-glycoprotein-like molecule. Biochem J 326:641–648
Ranieri-Raggi M, Martini D, Sabbatini ARM, Moir AJG, Raggi A (2003) Isolation by zinc-affinity chromatography of the histidine-proline-rich-glycoprotein molecule associated with rabbit skeletal muscle AMP deaminase. Evidence that the formation of a protein-protein complex between the catalytic subunit and the novel component is critical for the stability of the enzyme. Biochim Biophys Acta 1645:81–88
Ranieri-Raggi M, Moir AJG, Raggi A (2014) The role of histidine-proline-rich glycoprotein as zinc chaperone for skeletal muscle AMP deaminase. Biomolecules 4:474–497
Ronca F, Raggi A (2015) Structure-function relationships in mammalian histidine-proline-rich glycoprotein. Biochimie 118:207–220
Ronca F, Ranieri-Raggi M, Brown PE, Moir AJG, Raggi A (1994) Evidence of a species-differentiated regulatory domain within the N-terminal region of skeletal muscle AMP deaminase. Biochim Biophys Acta 1209:123–129
Ronca-Testoni S, Raggi A, Ronca G (1970) Muscle AMP aminohydrolase. III. A comparative study on the regulatory properties of skeletal muscle enzyme from various species. Biochim Biophys Acta 198:101–112
Rubio R, Berne RM, Dobson JG Jr (1973) Sites of adenosine production in cardiac and skeletal muscle. Am J Physiol 225:938–953
Rundell KW, Tullson PC, Terjung RL (1992a) AMP deaminase binding in contracting rat skeletal muscle. Am J Physiol 263:C287–C293
Rundell KW, Tullson PC, Terjung RL (1992b) Altered kinetics of AMP deaminase by myosin binding. Am J Physiol 263:C294–C299
Rundell KW, Tullson PC, Terjung RL (1993) AMP deaminase binding in rat skeletal muscle after high-intensity running. J Appl Physiol 74:2004–2006
Sabbatini ARM, Ranieri-Raggi M, Pollina L, Viacava P, Ashby JR, Moir AJG, Raggi A (1999) Presence in human skeletal muscle of an AMP deaminase-associated protein that reacts with an antibody to human plasma histidine-proline-rich glycoprotein. J Histochem Cytochem 47:255–260
Sabbatini AR, Toscano A, Aguennouz M, Martini D, Polizzi E, Ranieri-Raggi M, Moir AJG, Migliorato A, Musumeci O, Vita G, Raggi A (2006) Immunohistochemical analysis of human skeletal muscle AMP deaminase deficiency. Evidence of a correlation between the muscle HPRG content and the level of the residual AMP deaminase activity. J Muscle Res Cell Motil 27:83–92
Sabbatini ARM, Mattii L, Battolla B, Polizzi E, Martini D, Ranieri-Raggi M, Moir AJG, Raggi A (2011) Evidence that muscle cells do not express the histidine-rich glycoprotein associated with AMP deaminase but can internalise the plasma protein. Eur J Histochem 55:33–38
Sabina RL, Morisaki T, Clarke P, Eddy R, Shows TB, Morton CC, Holmes EW (1990) Characterization of the human and rat myoadenylate deaminase genes. J Biol Chem 265:9423–9433
Schmidt G (1928) Über fermentative desaminierung im muskel. Z Physiol Chem 179:243–269
Shiraki H, Miyamoto S, Matsuda Y, Momose E, Nakagawa H (1981) Possible correlation between binding of muscle type AMP deaminase to myofibrils and ammoniagenesis in rat skeletal muscle on electrical stimulation. Biochem Biophys Res Commun 100:1099–1103
Takeda S, Yamashita A, Maeda K, Maeda Y (2003) Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424:35–41
Tanokura M, Tawada Y, Ono A, Ohtsuki I (1983) Chymotryptic subfragments of troponin-T from rabbit skeletal-muscle. Interaction with tropomyosin, troponin-I and troponin-C. J Biochem 93:331–337
Tobacman LS (1988) Structure-function studies of the amino-terminal region of bovine cardiac troponin T. J Biol Chem 263:2668–2672
Tugues S, Roche F, Noguer O, Orlova A, Bhoi S, Padhan N, Akerud P, Honjo S, Selvaraju RK, Mazzone M, Tolmachev V, Claesson-Welsh L (2014) Histidine-rich glycoprotein uptake and turnover is mediated by mononuclear phagocytes. PLoS One 9:e107483
van Kuppevelt TH, Veerkamp JH, Fishbein WN, Ogasawara N, Sabina RL (1994) Immunolocalization of AMP-deaminase isozymes in human skeletal muscle and cultured muscle cells: concentration of isoform M at the neuromuscular junction. J Histochem Cytochem 42:861–868
Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ (2005) Ca(2+)-regulated structural changes in troponin. Proc Natl Acad Sci 102:5038–5043
Wakabayashi S (2013) New insights into the functions of histidine-rich glycoprotein. Int Rev Cell Mol Biol 304:467–493
Wang J, Jin JP (1997) Primary structure and developmental acidic to basic transition of 13 alternatively spliced mouse fast skeletal muscle troponin T isoforms. Gene 193:105–114
Wei B, Jin JP (2011) Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch Biochem Biophys 505:144–154
Wei B, Jin JP (2016) TNNT1, TNNT2, and TNNT3: isoform genes, regulation, and structure-function relationships. Gene 582:1–13
Westra HG, De Haan A, van Doorn H, De Haan EJ (1982) Short-term and persistent metabolic changes as induced by exercise. In: Addink ADF, Spronk N (eds) Exogenous and endogenous influences on metabolic and neural control. Pergamon Press, Oxford, pp 285–295
Wilson DK, Rudolph FB, Quiocho FA (1991) Atomic structure of adenosine deaminase complexed with a transition-state analog: understanding catalysis and immunodeficiency mutations. Science 252:1278–1284
Zhang Z, Biesiadecki BJ, Jin JP (2006) Selective deletion of the NH2-terminal variable region of cardiac troponin T in ischemia reperfusion by myofibril-associated μ-calpain cleavage. Biochemistry 45:11681–11694
Zhang T, Pereyra AS, Wang ZM, Birbrair A, Reisz JA, Files DC, Purcell L, Feng X, Messi ML, Feng H, Chalovich J, Jin JP, Furdui C, Delbono O (2016) Calpain inhibition rescues troponin T3 fragmentation, increases Cav1.1, and enhances skeletal muscle force in aging sedentary mice. Aging Cell 15:488–498
Zielke CL, Suelter CH (1971) Rabbit muscle adenosine 5′-monophosphate aminohydrolase. Characterization as a zinc metalloenzyme. J Biol Chem 246:2179–2186
Acknowledgements
The authors would like to thank Dr. Arthur J. G. Moir, Krebs Institute, University of Sheffield, UK, for the longtime cooperation and for critically reviewing this manuscript. This work was supported by Fondi di Ateneo, University of Pisa.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ronca, F., Raggi, A. Role of troponin T and AMP deaminase in the modulation of skeletal muscle contraction. Rend. Fis. Acc. Lincei 28, 143–158 (2017). https://doi.org/10.1007/s12210-016-0586-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12210-016-0586-7