
Abstract
Purpose
Shear-mediated thrombosis is a clinically relevant phenomenon that underlies excessive arterial thrombosis and device-induced thrombosis. Red blood cells are known to mechanically contribute to physiological hemostasis through margination of platelets and vWF, facilitating the unfurling of vWF multimers, and increasing the fraction of thrombus-contacting platelets. Shear also plays a role in this phenomenon, increasing both the degree of margination and the near-wall forces experienced by vWF and platelets leading to unfurling and activation. Despite this, the contribution of red blood cells in shear-induced platelet aggregation has not been fully investigated—specifically the effect of elevated hematocrit has not yet been demonstrated.
Methods
Here, a microfluidic model of a sudden expansion is presented as a platform for investigating platelet adhesion at hematocrits ranging from 0 to 60% and shear rates ranging from 1000 to 10,000 s−1. The sudden expansion geometry models nonphysiological flow separation characteristic to mechanical circulatory support devices, and the validatory framework of the FDA benchmark nozzle. PDMS microchannels were fabricated and coated with human collagen. Platelets were fluorescently tagged, and blood was reconstituted at variable hematocrit prior to perfusion experiments. Integrin function of selected blood samples was inhibited by a blocking antibody, and platelet adhesion and aggregation over the course of perfusion was monitored.
Results
Increasing shear rates at physiological and elevated hematocrit levels facilitate robust platelet adhesion and formation of large aggregates. Shear-induced platelet aggregation is demonstrated to be dependent on both αIIbβIII function and the presence of red blood cells. Inhibition of αIIbβIII results in an 86.4% reduction in overall platelet adhesion and an 85.7% reduction in thrombus size at 20-60% hematocrit. Hematocrit levels of 20% are inadequate for effective platelet margination and subsequent vWF tethering, resulting in notable decreases in platelet adhesion at 5000 and 10,000 s-1 compared to 40% and 60%. Inhibition of αIIbβIII triggered dramatic reductions in overall thrombus coverage and large aggregate formation. Stability of platelets tethered by vWF are demonstrated to be αIIbβIII-dependent, as adhesion of single platelets treated with A2A9, an anti-αIIbβIII blocking antibody, is transient and did not lead to sustained thrombus formation.
Conclusions
This study highlights driving factors in vWF-mediated platelet adhesion that are relevant to clinical suppression of shear-induced thrombosis and in vitro assays of platelet adhesion. Primarily, increasing hematocrit promotes platelet margination, permitting shear-induced platelet aggregation through αIIbβIII-mediated adhesion at supraphysiological shear rates.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The von Willebrand Factor (vWF) is an integral component of hemostatic regulation at high shear rates (> 1000 s−1) [1,2,3]. vWF is a globular multimeric glycoprotein present in blood acting as a mechanosensitive complex to permit binding of platelets and tethering to collagen [4, 5], as well as weaker interactions with other matrix proteins [6]. These functions are amplified by extreme shear rates (≥ 5000 s−1) [7, 8]. These supraphysiological shear rates exceed what is normally observed in the arterioles (1–2000 s−1) [1], and can be induced by pathology [7, 9] or mechanical circulatory support (MCS) devices [10, 11]. Flow disruptions caused by stenoses or MCS devices cause platelet activation and subsequent thrombosis [7,8,9, 12, 13] amplified by the concomitant upregulation of the platelet-capture function of vWF [7, 11]. In order to mitigate the occurrence of device-induced and arterial thrombosis, it is crucial to reduce platelet activation and aggregation through appropriate antiplatelet therapy. Additionally, it is imperative to gain a deeper understanding of the biochemical and mechanical factors responsible for vWF-mediated platelet capture and thrombus formation. Thus far, it has been demonstrated that thrombus deposition is exacerbated by both increasing shear rate [8, 14, 15] and rapid shear gradients [16, 17]. This phenomenon has a dual impact; firstly, high shear flows activate platelet and cause vWF stretching, and secondly, they promote platelet and plasma vWF margination, leading to increased concentration near the vessel wall [18], and thereby, enhancing their interaction with thrombi [19].
The margination of platelets, by increasing hematocrit proportionally, increases the near wall population of platelets [20], resulting in platelets experiencing higher shear forces than in the bulk flow. At physiological shear rates, this has been demonstrated in multiple platforms relevant to venous thrombosis or arterial hemostasis [19, 21]. This red blood cell-induced platelet margination amplifies the shear forces experienced by platelets and enhances subsequent tethering by vWF [17, 22]. The effects of shear rate gradients on vWF adhesion have been studied directly in pure extensional flow [23,24,25], at the outlet of a severe stenotic region [9], and indirectly in a canonical model of device-induced thrombosis [26]. Previous work with centimeter-scale models of in vitro [27] and in silico [28, 29] backwards-facing step (BFS), or sudden expansion models, as well as microfluidic models of device-relevant geometric flaws [30,31,32] influenced the design presented here, a microfluidic model of a sudden expansion with a sub-millimeter backwards-facing step geometry.
On a clinical level, heightened hematocrit levels have been linked to a greater incidence of infarction, deep venous thrombosis, and stroke [33]. Notably, elevated hematocrit levels are recognized for their role in facilitating increased platelet margination [34]. Recent studies have revealed that the presence of red blood cells simultaneously promotes the margination and unfurling of vWF multimers [17], further contributing to the complex dynamics of platelet-vWF interactions. In a model of venous thrombosis, Lehmann et al. [21] demonstrated a reliance on margination for the formation of stable thrombi, by way of red blood cell margination of platelets into the valve pocket region; in this case the binding receptor GPVI was found to be responsible for tethering and subsequent platelet activation at this physiological shear range (10–500 s−1). A study of arterial thrombosis produced similar results, in which a microfluidic in vitro and murine in vivo model (750–1000 s−1) demonstrated enhanced platelet accumulation and rapid thrombus formation as a result of elevated haematocrit [19, 21]. However, neither of these studies investigated whether this phenomenon was a driving factor at supraphysiological shear rates relevant to pathological and MCS flow. At these supraphysiological shear rates (~ 10,000 s−1), GPIb-vWF interactions have been suggested to be the driving factor behind shear-induced platelet aggregation (SIPA) [35], while the action of red blood cells was deemed a secondary contributor [7, 36]. Recently, in vitro and in silico models of initial platelet adhesion have highlighted that platelet tethering by vWF through GPIb [37,38,39] is followed by the subsequent activation of αIIbβIII. These studies highlight a potential avenue for prevention of SIPA through targeting of mechanisms governing microthrombus formation. SIPA’s impact on arterial thrombosis influenced this investigation into how hematocrit (HCT) may facilitate shear rate effects on platelet capture, and how inhibition of vWF-αIIbβIII interactions affects microthrombi stabilization. The primary hypothesis of this study is that platelet aggregation induced by supraphysiological shear rates is a hematocrit-dependent process mediated by αIIbβIII receptor adhesive interactions. To this purpose, we have used a microfluidic sudden expansion of 90˚, which has been demonstrated previously to mediate platelet adhesion [21, 26,27,28,29,30, 32] due to the fluid mechanics imposed by this geometry.
Methods
Blood Collection and Preparation
Human blood was acquired from healthy donors through a Penn State IRB approved protocol for venipuncture. Donors were comprised of 8 females and 10 males (aged 18–40, mean 22.4 years), who had not taken any antiplatelet or anti-inflammatory medication for at least 5 days preceding blood collection. Mean platelet concentration was 299 ± 87 *106/mL of plasma. For each condition, at least one experiment was performed for each donor listed. The summary of donors for each experimental condition is outlined in Table 1. Whole blood was drawn into a blood bag (Terumo, Tokyo Japan) prefilled with 3.2% sodium citrate, in a ratio of 9:1 (final anticoagulant concentration was 0.32% sodium citrate). Blood was separated into packed red blood cells and platelet-rich plasma (PRP) by spinning at 300×g for 30 min at 25 °C. The upper portion of PRP was removed, and a small aliquot was set aside for platelet counting. The red blood cells and buffy coat then underwent a second spin at 3600 rpm for 30 min at 25 °C. The buffy coat and platelet poor plasma was discarded, and packed red blood cells were transferred to a new conical tube. The separated PRP was incubated on a rocker for 30 min at 37 °C with lipophilic dye 3,3ʹ-dihexyloxacarbocyanine iodide (DiOC6, 1 μM) for platelets before reconstitution to the desired hematocrit (0, 20, 40, 60%), verified by centrifugation in an Autocrit Ultra3 (Clay Adams, Parsippany NJ). Unstained platelets aliquoted from the separated PRP were counted within a hemocytometer. Platelet count was recorded and used to normalize microscopy results. During analysis, the approximate mean platelet concentration (300*106/mL) was divided by the platelet count of each donor to obtain individual correction factors. The metrics of platelet adhesion/thrombus growth for each donor were then multiplied by this correction factor to determine if platelet concentration was a significant factor. To inhibit the function of the αIIbβIII integrin, a monoclonal CD41/CD61 antibody was obtained commercially (BioLegend, San Diego CA, cat#359802, A2A9/6 clone). For these αIIbβIII-inhibited samples, separated PRP was incubated at 37 °C with A2A9 [40, 41] at a concentration of 5 µg/mL for 30 min immediately following the DiOC6 incubation. To differentiate the effects of αIIbβIII-inhibition from the contribution of the GPIb receptor and red blood cell secretions, auxiliary experiments were conducted in which GPIb was blocked, or ADP-depleted ghost red blood cells were used in reconstitution (Supplementary Information). To maintain equivalent bulk platelet concentrations, lower HCT (0–40%) samples were buffered with HEPES-buffered saline (140 mM NaCl, 1.5 mM Na2HPO4•2H2O, 50 mM HEPES) [42] to maintain 40% v/v PRP composition. Reconstituted blood was placed on a rocker (Fisher Scientific, Waltham MA) at 37 °C until needed for perfusion. To determine whether increased shear stress was a determining factor in increased thrombus deposition, PRP-glycerol mixtures were created to match the viscosities of 20, 40, and 60% HCT blood samples (2.3, 3.9, 8.24 cP, respectively). HEPES-buffered saline and glycerol aliquots were warmed to 37 °C on an incubated rocker prior to blood reconstitution. All reconstituted blood samples were brought to and maintained at 37 ˚C until perfusion. The shear stresses associated with these viscosities at each shear rate are listed in Supplemental Table 1. Similarly, these blood analog samples were incubated with 1 μM DiOC6, buffered with HEPES-buffered saline to maintain 40% v/v PRP composition and kept at 37 °C until perfusion. All adhesion assays were conducted within 6 h of blood donation.
Microchannel Fabrication
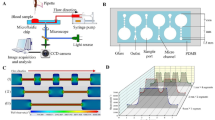
Polydimethylsiloxane (PDMS) microchannels were fabricated, serving as a testing platform for shear-altered deposition. Master molds were designed in Fusion 360 (Autodesk, San Rafael CA) and SU-8 photoresist coated following standard photoresist lithography procedures [43]. Sylgard 184 Silicone (Ellsworth Adhesives, Irondequoit NY) was mixed in a 10:1 base to curing agent ratio. The elastomer components were mixed, degassed for 20 min, and allowed to cure for 4 h at 60 °C following established soft lithography protocols [15]. A sudden expansion (SE) geometry (Fig. 1A–B) was chosen to evaluate the effects on shear-mediated adhesion. Variations of this geometry have been studied in literature as representative of device geometries [31], or roughly analogous to the outlet region of a stenosis [9]. The SE fluid dynamics are characterized by reduced flow immediately distal to the step, with a region of flow separation at higher Reynolds numbers (Supplemental Fig. 1). The flow parameters governing these experiments are summarized in Table 2. The geometric features of the channel can be observed in Fig. 1A, with regions of interest denoted in Fig. 1B. After curing, PDMS chips were peeled from the SU-8 master and cut to fit glass slides. Inlet and outlet holes were punched with a 1 mm biopsy punch (Integra, Princeton NJ). The peeled PDMS chips were rinsed three times with 70% ethanol (EtOH) and dried with compressed air. Glass microscope slides were cleaned in a similar fashion. Next, PDMS chips were sealed to glass microslides using a plasma cleaner (Harrick Plasma, Ithaca NY) at 18W, ~ 550 mTorr, for 45 seconds. Sealed channels were then placed on a hot plate at 80 °C for 20 min to catalyze the bonding reaction and form a permanent seal. After cooling, the sealed microchannels were rinsed with 100% EtOH, followed by 1X PBS.

A Schematic of SE channel dimensions. Channels are 50 μm tall in the z-direction. Figure is not drawn to scale. B Regions of interests (ROIs) in processing surface deposition. SE represents the entire field of view. Q1, Q2, and Q3 are the region directly prior to the expansion, the region following the expansion, and the step region, respectively. C Blood perfusion through microfluidic chip. D Immunofluorescent microscopy schematic
Microchannel Preparation
Following the fabrication process, channels were coated with human type I collagen (Advanced Biomatrix, Carlsbad CA) at 100 µg/mL to create an adhesive surface. Collagen was diluted from 3.2 mg/mL in ultrapure deionized water with 5 μL droplets of 0.1M NaOH added slowly to bring the pH to 7.0 [44]. Prior to collagen functionalization, channels underwent a washing cycle of perfused 100% ethanol, 1X PBS, and ultrapure water. Following the washing cycle, channels were incubated with collagen for 2 h at room temperature while covered. After this passivation period, channels were again rinsed with ultrapure water and stored overnight at 4˚C until use. The collagen coating was labeled and imaged to ensure a homogenous surface (Supplementary Information). Channels were brought to room temperature before perfusion experiments. Twenty cm lengths of 0.79 mm inner diameter (I.D.) Tygon tubing (Grainger, Lake Forest IL), 3 mL syringes (8.65 mm I.D.), and 0.79 mm I.D. barbed luer connecters (Qosina, Ronkonkoma NY) were assembled and incubated with 2% bovine serum albumin overnight to prevent interior platelet adhesion. These syringes were emptied, rinsed, and filled with blood prior to connection to the punched inlet hole (Fig. 1C–D).
Computational Fluid Dynamics
Shear rates were calculated from Eq. (1), where Q is the volumetric flow rate, h is the channel height, and w is the channel width. Velocity profiles were calculated in an ANSYS FLUENT (ANSYS, Canonsburg PA) computational simulation and verified by acquiring velocity data with a micro-PIV (particle image velocimetry) system (TSI Inc., Shoreview MN). Shear rates were chosen to represent healthy arterial (1000 s−1), stenotic (5000 s−1), and device-relevant (10,000 s−1) shear rates (Table 2), corresponding to the experimental flow rates of 5, 25, and 50 μL/min. The shear rate for each case is denoted by the average shear rate of the upstream portion (cross-section of 200 × 50 μm) of the backwards-facing step. The step region was the site of rapid deceleration, followed by a downstream region with an average shear rate half that of the upstream portion. Blood was perfused through the microchannels with a programmable syringe pump (Harvard Apparatus, Holliston MA) at volumetric flow rates chosen to achieve the desired shear rates.
Experimental Fluid Dynamics Design
Fabricated SE channels were mounted on a microscope stage and taped in place. Platelet rich plasma (PRP, 0 HCT%), and blood at 20, 40, and 60% HCT were perfused over 100 μg/mL collagen for 5 min at 1000, 5000, and 10,000 s−1. For each HCT and shear rate condition, both unmodified control blood samples and A2A9-blocked samples were perfused. In total, 18 blood donors were used in this study.
Micro-PIV was performed using a Nikon Eclipse TE300 inverted fluorescent microscope (Nikon, Melville NY) with a Plan Fluor 20X (Nikon, Melville, NY) objective, while images were captured with a Powerview 4MP-HS CCD camera (TSI Inc., Shoreview MN). One hundred image pairs were captured for each shear rate before processing in Insight 4G (TSI Inc., Shoreview MN). Polysterene microparticles (Thermo Fisher Scientific, Waltham, MA) of 1 μm diameter and 540/560 nm excitation/emission spectra were seeded at 2% concentration in ultrapure water and excited with an Nd:YAG 532 nm laser (New Wave Research, Inc., Fremont, CA). Real-time immunofluorescence images were acquired during perfusion with an Olympus inverted microscope and CMOS camera (Olympus, Tokyo Japan) at a rate of 1 frame per second and an exposure time of 100 ms. A 20x/0.40 objective lens was used with a pixel resolution of 0.293 μm/pixel. The corner of the step was moved to the center of the field of view using the digital reticle tool in CellSens (Olympus, Tokyo Japan), to ensure simultaneous image capture of all regions of interest. A schematic of the microscopy experimental setup is shown in Fig. 1C with the light path through the microfluidic device illustrated in Fig. 1D.
Image Analysis, Statistics
Images were processed with a custom FIJI macro (NIH, Bethesda MD) and MATLAB (Mathworks, Natick MA). Fluorescent images were converted to binary, and SAC was quantified. Channels were divided into quadrants for processing (Fig. 1B). Quadrants were chosen to evaluate the effects of flow deceleration at the step. Using the corner of the step as a reference point, quadrants were digitally delineated as regions of interest in FIJI, and processed individually. For every frame of each region of interest, the “Analyze Particles” tools was used to acquire the number of thrombi, total thrombus area, mean thrombus area (MTA), and % surface area coverage (% SAC). Statistics were analyzed in GraphPad Prism (GraphPad, San Diego CA) using two-way ANOVA, Kruskal-Wallis, and Holm-Sidak or Mann-Whitney U tests where appropriate. Significance is presented as n.s. (p > 0.05), * (p < 0.05), ** (p < 0.01), *** (p < 0.001), or **** (p < 0.0001).
Results
Effect of Hematocrit and Shear Rate on Platelet Deposition
Hematocrit (HCT) was observed to have a significant effect on platelet deposition and morphology, particularly in conjunction with high shear rates. Representative images of the control conditions investigated in the SE channel are shown (Fig. 2), in which it can be observed that deposition follows the streamlines (Supplemental Fig. 2) within the step region. This distal step region (Q3) promoted platelet adhesion due to flow separation and deceleration, as observed in the representative image for 60% HCT at 5000 s−1 (Fig. 2). This region typically the site of highest platelet adhesion, for both control and A2A9-modified samples. Representative images of A2A9-modifed samples are shown in Fig. 3. The step feature itself acted as a nidus for initial platelet adhesion and subsequent aggregate formation, as seen at 10,000 s−1 for both representative 60% HCT images (Fig. 2-3). The corner was the site of the sharpest shear rate gradient, promoting rapid, localized aggregate formation in 50–85% of control samples and, to a lesser degree, some A2A9-modified samples. Hematocrit was observed to have a notable effect on platelet deposition that was compounded by deceleration of flow (Supplemental Table 3). SAC was assessed in each image frame, illustrating thrombus growth rates for all conditions (Fig. 4). At 5000 and 10,000 s−1, adhesion of control samples rapidly exceeded adhesion of A2A9 modified samples within the first 30–60 s of perfusion. At 1000 s−1, the gap in adhesion rates between control and A2A9-modified samples was less evident within this time frame. As SAC surpassed 10% coverage, embolization became more common (Fig. 4) due to the greater drag profile of larger thrombi. Values used to compare adhesion between conditions and statistically analyze data were taken from the final frame of acquisition (Fig. 5–7).

Representative images of control (unmodified by A2A9) blood sample deposition after 5 min of perfusion. Dotted red lines denote channel boundaries. N = 7-9, n = 9-14. Scale bar = 50 μm

Representative images of A2A9-modified (αIIbβIII-inhibited) blood sample deposition after 5 min of perfusion. Dotted red lines denote channel boundaries. N = 6, n = 6-11. Scale bar = 50 μm

Timelapse of platelet deposition in each processing ROI over 5 min of perfusion. Columns separate data by shear rate (1–10,000 s-1) while rows denote processing ROI (Q1-3). Each window shows the SAC (%) for the four HCT levels with both control and A2A9-modified samples. Images taken at 1 frame per second. Data are presented as mean ± SEM

SAC (%) in each processing ROI after 5 min of perfusion of control (left column) and A2A9-treated (right column) samples. Data are presented as mean ± SEM. Significance of increasing hematocrit as a predictive variable was assessed at each shear rate by individual Kruskal-Wallis tests within groups. Inset boxes display results of two-way ANOVA assessment for each quadrant, in which shear rate and hematocrit are the independent variables. Significance is presented as *(p < 0.05), **(p < 0.01), ***(p < 0.001), or ****(p < 0.0001), or n.s. (not significant if not denoted).
Overall adhesion of platelets (SAC) within the collagen coated SE channel was affected by hematocrit at all shear rates, and the increase in deposition was most pronounced in Q3 (Fig. 5). Control blood samples at 20, 40, and 60% HCT all displayed shear-dependent increases in surface coverage and MTA. Inhibition of αIIbβIII by A2A9 incubation resulted in reduced platelet deposition for all conditions. A two-way ANOVA analysis indicated that both hematocrit and shear rate were significant factors in increased MTA for all control conditions, in all regions of interests (Fig. 6, Supplemental Table 2). Hematocrit was a significant factor for all quadrants in driving increased SAC, while shear rate was a significant factor only in Quadrant 3 (Fig. 5). Hematocrit was strongly correlated (p < 0.0001) to increased thrombus formation of unmodified blood samples for all regions of interest. Shear rate was significant in Q3 and was still a contributor to increased deposition in Q1 and Q2, while not reaching statistical significance (Fig. 5). For A2A9-modified samples, hematocrit was also a significant factor in increasing platelet deposition, albeit to a lesser degree. Sixty percent HCT permitted moderate platelet deposition in spite of the A2A9 treatment, suggesting a weakening of the blocking effects on platelet adhesion and aggregation when opposed by elevated HCT. Conversely, shear rate was also significant in that a negative correlation was observed in 0, 20, and 40% HCT, with no correlation seen at 60% HCT. Kruskal-Wallis tests were subsequently performed to confirm these findings. The significance of increasing hematocrit as a predictive variable for each shear rate can be found in Fig. 5. Pairwise comparisons between conditions can be found in the attached Supplementary Information. Shear rate was found to only be statistically significant for deposition of control samples at 40 and 60% HCT in Q3. For A2A9-modified samples, inverse correlations with shear for 20 and 40% were found to be statistically significant in Q1 and Q2. Pairwise comparisons between control and A2A9-modified samples were made by the Mann-Whitney U test. While platelet deposition for control samples was consistent at all conditions, statistically significant differences were observed primarily in 40 and 60% HCT conditions. Notably, statistical significance was not achieved in pairwise comparisons at 1000 s−1 in the Q3 region for 40 and 60% HCT.

Mean thrombus area (μm2) in each processing ROI after 5 min of perfusion of of control (left column) and A2A9-treated (right column) samples. Data are presented as mean ± SEM. Significance of increasing hematocrit as a predictive variable was assessed at each shear rate by individual Kruskal-Wallis tests within groups. Inset boxes display results of two-way ANOVA assessment for each quadrant, in which shear rate and hematocrit are the independent variables. Significance is presented as *(p < 0.05), **(p < 0.01), ***(p < 0.001), or ****(p < 0.0001), or n.s. (not significant if not denoted)
Increasing hematocrit and increasing shear rate both resulted in the formation of large platelet aggregates, as measured by MTA. Increasing hematocrit resulted in larger thrombus surface deposits in control blood samples (Figs. 6, 7). SIPA behavior was observed at 20, 40, and 60% HCT. A two-way ANOVA assessment was used to assess the effect of hematocrit and shear rate on thrombus morphology (Supplemental Table 2). For control samples, hematocrit was a significant factor in MTA, particularly in Q3 (Fig. 6). However, contrary to thrombus SAC, MTA was affected primarily by increasing shear rate, with the greatest effect seen in Q3 (p < 0.0001). For A2A9-modified samples, hematocrit and shear rate were significant factors only in the Q3 region. SIPA behavior was still observed in A2A9 samples, but only for 40 and 60% HCT blood and to a lesser degree than in control samples (Fig. 7). Visually, thrombus growth occurred preferentially along streamlines as platelets were added to the distal ends of adherent microthrombi. Deposition also occurred frequently at the step for control samples (Fig. 8). Again, internal Kruskal-Wallis tests were performed to confirm these findings. The significance of HCT at each shear rate can be observed in Fig. 6. Shear rate was found to be a statistically significant factor in MTA of control samples at 20, 40, and 60% HCT for all regions of interest. In A2A9 samples, shear rate was significant only for 40 and 60% HCT in Q3. Pairwise comparisons between control and A2A9-modified samples were made by the Mann-Whitney U test (Supplementary Information). Statistically significant differences in MTA between control and A2A9-modified samples were observed for 20, 40 and 60% HCT for all conditions.

Heatmap representation of SAC (%) and mean thrombus area (μm2) of unmodified and A2A9-modified thrombi after 5 min of perfusion. Surface` area coverage was calculated in each ROI, Q1 (Upper left boxes), Q2 (upper right boxes), Q3 (lower right boxes), for each condition. Mean values are shown, color is scaled to highest value across all 24 conditions

Deposition frequency across all conditions at 40% HCT. Frequency maps are constructed from overlaid binary images of the final frame after 5 min of perfusion. After filtering, processing, and thresholding in FIJI, individual binary images were overlaid and converted into a stack by using the step as a reference. Average intensity was then calculated to obtain frequency values. Calibration bar for frequency is found on the left of the image. Red lines denote channel boundaries. Scale bar = 50 μm
Effect of Shear Rate Gradient on Platelet Deposition
The sudden expansion model was divided into three processing regions to assess the relationship between local fluid dynamics and platelet deposition. The three quadrants (Q1, Q2, and Q3) each had different velocity profiles (Supplemental Fig. 1, S3). The velocity profile in Q1 is similar to Poiseuille flow in a straight channel. Q2 is characterized by a moderate, nearly linear deceleration as the velocity approaches 50% of the upstream velocity. Q3 is the most complex flow, with a rapid zone of deceleration in the corner pocket region furthest from the step, and more moderate negative shear gradients towards the center of the channel (Supplemental Fig. 4, Supplemental Table 3). Kruskal-Wallis analyses did not reveal any significant differences between these quadrants, although trends were apparent. Platelet deposition (% SAC) tended to be highest in Q3 for both control and A2A9-modified samples (Figs. 4, 5), and HCT-mediated deposition of A2A9-modified samples at 5–10,000 s−1 was significant only in Q3 (Fig. 5, Supplemental Table 2). While quadrant selection seemed to play no role in MTA for control samples, larger thrombi did tend to form in Q3 for A2A9-modified samples (Figs. 6, 7). To provide a more detailed depiction of thrombus formation sites in the channel, a frequency map of deposition for all 40% HCT conditions is provided (Fig. 8).
Effects of Margination vs. Increased Shear Stress
To further support that the hematocrit-dependent increases in thrombus formation were tied to platelet margination and not entirely a result of increased shear stress (Supplemental Table 1) due to corresponding increases in viscosity, control experiments were performed using PRP-glycerol analogs. Perfusion of these 20, 40, and 60% HCT analog mixtures showed no increase in platelet adhesion relative to 0% HCT samples (Supplemental Fig. 5).
Assessment of Donor Variability
In order to account for variability between donors, data were normalized to a platelet concentration of 300 * 106 /mL from the recorded concentrations for each donor. Individual data from experiments run for each donor were “platelet-corrected” and compared (Supplemental Fig. 6). No significant differences were detected in pairwise comparisons of raw and platelet-corrected data. Data from male and female donors were separated and compared to detect any sex-specific differences (Supplemental Fig. 7). Pairwise comparisons revealed slightly higher platelet adhesion on average for male donors than female donors, but none of these differences reached statistical significance. Male thrombi were also larger on average than those of female donors, with differences of 6.9, 78.2, 86.8, and 41.1 % at 10,000 s-1 for 0, 20, 40, and 60% HCT control samples, respectively. These differences were statistically significant for 20 and 40% HCT. Despite these differences, the observed increases in SAC and MTA as a result of increasing HCT and shear rate remained statistically significant for both male and female donors.
Investigation of Additional Biochemical Mechanisms of Aggregation
To ensure that the observed SIPA and increased platelet adhesion was due to mechanical contributions of red blood cells mediated by αIIbβIII, experiments blocking the function of the GPIb receptor and red blood cell secretion of ADP were conducted (detailed methodology available in Supplementary Information). ADP-depleted ghost red blood cells were used to reconstitute blood at 20-60% HCT, displaying comparable adhesion to unmodified control samples. There were no significant differences in SAC between ghost cell and control samples at any HCT, shear rate, or within any region of interest (Supplemental Fig. 10). Modestly decreased aggregation was observed, though a statistically significant reduction in MTA was detected only for 40% HCT at 5000 s-1 in the Q1 region. For all other conditions, aggregation of ghost cell samples was either significantly higher than that of control samples (60% HCT at 1000 s−1, in Q1 and Q3 regions), or not significantly different (Supplemental Fig. 11). In GPIb-blocked samples, SAC was significantly reduced for all conditions at shear rates exceeding 1000 s−1 (Supplemental Fig. 10). Platelet adhesion was notably absent at these shear rates. Aggregation was also absent, except for the Q3 region. MTA of control and GPIb-blocked samples were comparable for both 40 and 60% HCT at 5000 s−1, and 60% HCT at 10,000 s−1 (Supplemental Fig. 11). This aggregation occurred despite systemic loss of platelet adhesion, and in the same location within the step of the sudden expansion (Supplemental Figs. 12–14).
Discussion
Microfluidic flow chambers have become ubiquitous in conducting assays of platelet adhesion and clot formation. Here, we modeled a sudden expansion to induce flow separation and nonphysiological flow patterns observed in mechanical circulatory support devices [28,29,30, 32] and used real-time immunofluorescent microscopy to capture platelet adhesion. This geometry introduced flow perturbations that are characteristic to benchtop [27, 30, 32] and numerical [10, 26, 28, 29, 48] models of device-induced thrombosis. The negative shear gradient experienced by platelets at the expansion was a nidus for clot formation, approximating the disturbed flow of device-induced thrombosis in a more relevant way than the idealized parallel plate chamber. The most severe shear rate gradient was imposed at the step feature itself—leading to localized aggregate formation visible in representative images (Figs. 2, 3) and deposition map (Fig. 8). This shear rate gradient promotes rapid unfurling of vWF multimers due to increased intra-multimer tension [25]. This sharp spike in shear rate at the step followed by deceleration in Q3 (Supplemental Fig. 4) mediates localized aggregation through rapid unfurling and increased adhesion to collagen. While feature-induced adhesion has been observed in previously discussed models of 90˚ expansions, this binding behavior has not previously been tied to αIIbβIII function. Flow rates were prescribed to approximate the shear forces experienced by platelets in arterial, pathological, and device-relevant flow environments. A homogenous collagen surface [44] was used as a proven adhesive substrate for platelet adhesion. Reconstituted blood of variable hematocrit was perfused in this model to determine the effect of hematocrit on platelet adhesion and aggregation mediated by the αIIbβIII integrin. While red blood cells are known to play a critical role in platelet-wall transport, the effect of elevated hematocrit at supraphysiological shear rates has not previously been demonstrated. The importance of αIIbβIII is highlighted to play a crucial and previously uninvestigated part in shear-induced platelet aggregation, where vWF is the primary ligand rather than fibrinogen. The antiplatelet effect of a known aggregation inhibitor was modestly neutralized by elevated hematocrit and a region of flow deceleration that increased the rate of platelet capture.
At supraphysiological shear rates, large, platelet-rich thrombi form due to vWF-mediated platelet adhesion, and subsequent activation [38] and shear-induced platelet aggregation, or SIPA [7] mediated by GPIb. Thus far, the roles of erythrocyte-induced platelet margination and the αIIbβIII integrin have yet to be defined within the realm of SIPA. In this study, the effects of supra-physiological shear rate, hematocrit, and the contribution of αIIbβIII have been investigated. At all shear rates, the need for red blood cells to facilitate platelet adhesion through margination was further validated in support of previous studies [19, 21, 49, 50]. Elevated hematocrit increased not only the quantity of adherent platelets but also the likelihood of aggregation to an existing microthrombus. Elevated hematocrit can result from congenital heart defects, dehydration, or diseases such as polycythemia vera (PV) [51]. Conversely, reduced hematocrit, approximated by the 20% HCT condition, has been documented in anemic and VAD patients [52], and those with blood or bone marrow disorders. Long et al. describe reduced hematocrit of pediatrics ranging from 20 to 60% [53]. Large variance in hematocrit has been recorded due to these conditions as well as a result of clinical intervention such as through infusions, or device-induced blood damage. Adhesion at 20% HCT was notably reduced compared to the physiological (40%) HCT condition, and the elevated (60%) HCT condition slightly increased platelet surface coverage. Alternatively, blood samples modified by monoclonal antibody A2A9 displayed neutered adhesive behavior as a result of αIIbβIII inhibition. The A2A9 antibody competitively inhibited [40] αIIbβIII binding sites for the arginylglycylaspartic acid (RGD) binding motifs present on vWF [54] and fibrinogen [55]. For A2A9-modified blood samples, increasing hematocrit still resulted in increased surface coverage by adherent platelets. This was particularly evident when HCT increased from 40 to 60%, and in the steep deceleration of flow in Q3, both modestly compensating for αIIbβIII blocking relative to lower HCT or other regions.
The results of this work are similar to those reported by Chen et al. [50], in that shear-mediated adhesion was permitted only by 40 and 60% HCT. However, key differences distinguish the two studies. First, the adhesive substrate used here was immobilized collagen rather than immobilized vWF, requiring localized adhesion, unfurling, and platelet tethering to occur prior to extensive thrombus formation. Second, Chen et al. inhibited the activation and aggregation responses of platelets, studying only GPIb interactions with immobilized vWF. This resulted in only singular platelet adhesion, without the formation of microthrombi. The work presented here is more comparable to the experiments performed by Spann et al. [49], in which the authors investigated the effect of HCT on vWF-GPIb and vWF-αIIbβIII adhesion to collagen. While this study [49] provided valuable insight into mechanisms of arterial hemostasis, the lack of supraphysiological shear rates investigated highlighted a gap in knowledge as to whether this mechanism was relevant to SIPA. The study presented here aimed to address this less commonly investigated flow condition—investigating both platelet adhesion and microthrombus formation induced by elevated hematocrit at shear rates surpassing those found in arterial circulation. Elevated hematocrit caused an increase in platelet aggregation that was not described in the works of Vignoli et al. [51], which studied the effects of PV on platelet adhesion at arterial shear, or in Walton et al. [19], which investigated rates of arterial thrombus formation in murine and in vitro microfluidic models. While these studies effectively demonstrated a dependence on red blood cells for effective platelet margination and adhesion, little consideration was given to the contribution of red blood cells to SIPA. Additionally, the few studies that have investigated the effect of hematocrit in a system of disturbed flow have not reported the effects on thrombus formation [56] or have focused on physiological shear rates [21].
The molecular events preceding SIPA have been studied extensively [25, 50, 57, 58], and recent work has investigated SIPA formation at multiple scales [7, 8, 13, 35, 36]. vWF-GPIb bonds have been highlighted as a primary mechanism of SIPA, as high shear flow elongates vWF multimers and encapsulates platelets [35]. In this platform, these vWF-GPIb bonds in the absence of αIIbβII function were insufficient to mediate notable increases in microthrombus size, or elevated platelet adhesion, contrasting with recent conclusions postulated by Liu et al. [7, 35]. While these works demonstrated an activation-independent reliance on GPIb for rapid platelet adhesion and recruitment, the extremely short time scale (~10ms) limited understanding of the SIPA-related mechanisms beyond microthrombus initiation. Later stage aggregation and resistance to embolization over 5 min of perfusion in this system were shown to not only be αIIbβII-dependent but enhanced by flow deceleration at the step. This is more in line with the thrombus growth demonstrated by the same group [13] at a larger scale, in which longer time scales permitted large, vWF-rich thrombus formation at the outlet of a stenosis. By calculating both SAC and MTA, trends in binding behavior were observed that could not occur within the millisecond timeframe of initial platelet tethering reported in previous studies. SAC measured the overall adhesion of platelets to the channel, whether to collagen or to existing thrombi. MTA was used to differentiate between these two binding locations—larger thrombi indicated aggregation while smaller thrombi indicated primarily platelet adhesion to collagen. The results presented here demonstrate that this is made possible through integrin binding following the prerequisite initial tethering of platelets through vWF-GPIb bonds.
In this model, increasing hematocrit significantly increased deposition of platelets onto collagen (100 µg/mL) at shear rates of 1000, 5000, and 10,000 s−1. Platelet deposition at 0% HCT was negligible, while increases in deposition were observed when HCT increased from 20 to 60%, particularly in Q3, the step region. Increasing hematocrit and shear rate also influenced thrombus morphology and size, as measured by MTA. Platelets that initially adhered to collagen were individually resolvable (6–8 pixels), but these changes in binding behavior were characterized by overall trends within the system. At higher shear rates and hematocrit, large thrombi grew rapidly in separated pockets. These contiguous deposits are visible in both representative images (Figs. 2, 3) and the deposition frequency map (Fig. 8). At arterial shear rates and reduced hematocrit, thrombi were more evenly distributed but much smaller, reflected in the reported MTA. This change in MTA highlights a shear threshold at which platelet binding shifts primarily from platelet-collagen bonds to platelet-platelet bonds. The shear range at which this occurs is in agreement both with experimental observations of SIPA [7, 9, 59, 60] and molecular studies of vWF-unfurling thresholds [4, 17, 57, 61], further supporting that shear-induced thrombi are vWF-rich in nature [13]. This ties closely to conditions observed in MCS devices, where high shear rates cause vWF-mediated platelet adhesion to be dominant [11]. In A2A9 experiments this effect was greatly reduced, although a modest shear-related increase in MTA was observed for 40 and 60% HCT in the Q3 region. SIPA mediated by short-lived GPIb bonds in this region might be made possible due to longer residence times for lower shear rates, increasing the likelihood of microthrombus stabilization. For A2A9-modified samples perfused at 5000 and 10,000 s−1, HCT played a significant role in only the Q3 region. At 1000 s−1, HCT affects adhesion in the Q2 region, which aligns with the more gradual flow deceleration in this quadrant relative to Q3.
Increasing shear rate was negatively correlated to deposition of the A2A9-modified samples, particularly in the Q1 region where there was no flow deceleration. Increasing shear rate decreased the platelet-collagen exposure time and thus the likelihood of permanent adhesion. Transient adhesion, translocation, and subsequent embolization of individual platelets were observed during real-time imaging of A2A9 experiments (Video available in Supplementary Information). This is congruent with understanding of the GPIb receptor [4, 7, 22, 35], which mediates short-lived bonds through vWF tethering and platelet rolling. When the function of the αIIbβIII receptor was blocked, flow-induced activation did not induce platelet spreading and thrombus stabilization. Some platelets were able to adhere permanently over the course of the experiment, likely due to GPVI bond stabilization [21, 62]. Increasing HCT increased the number of platelets that did so, due to an increase of the near-wall platelet population [19, 63]. In the Q3 region, the steep deceleration relative to the other quadrants increased the platelet-wall contact time, and thus, the likelihood of stabilization.
Sudden 90° expansions characteristic to device-induced thrombosis [26, 28, 29] or the distal outlet of a stenosis [16, 59], have been used previously to identify shear and transport conditions favorable to platelet adhesion [21]. Within the SE model presented here, the effects of increasing shear rate and HCT for both SAC and MTA were enhanced in the Q3 region, for both unmodified and A2A9-modified samples. The negative shear rate gradient imposed within the expansion decelerated flowing platelets, increasing exposure time and subsequent adhesion. While HCT-mediated deposition in all control experiments was increased, more might be gleaned by observing the adhesive behavior of the A2A9-modified platelets. Due to αIIbβIII inhibition, platelet adhesion at these shear rates is dependent primarily on short-lived vWF-GPIb interactions [63]. Increasing HCT had the dual effect of enhancing the platelet population that might be tethered transiently by vWF and increasing the contact time with existing thrombi to permit stabilization [19]. This was compounded by deceleration in the Q3 region, which permitted both HCT-enhanced platelet margination to the step corner and a higher residence time, resulting in adhesive behavior similar to that observed in other in vitro models of 90° expansions [21, 26, 28, 30]. As a result, a HCT-dependent increase was observed for A2A9-modified samples at 5000 and 10,000 s-1 in the Q3 region. Similarly, shear rate was strongly correlated with increased SAC of control samples for 40 and 60% HCT samples and in the Q3 region. Given long enough formation times and adequate shielding from embolization-inducing hydrodynamic drag, vWF-GPIb bonds alone might be sufficient for SIPA, as reported by Kim et al. [13]. Deposition within the BFS model was observed to follow flow streamlines for both control and A2A9 experiments. Figure 8 provides further context by overlaying binary images of all experiments at 40% HCT to produce a representative frequency map. To confirm that this was not due to a realignment of collagen induced by high shear flow, validation images were taken in separate channels after staining for collagen. No notable change in the fluorescent intensity of the labeled collagen film was observed as a result of perfusion, and the film remained homogenously distributed (Supplemental Figs. 8–9).
The inclusion of a 60% HCT condition was useful as an assessment of the effect of elevated HCT over the physiological norm on platelet adhesion. Here, we observed that 60% HCT did not significantly increase platelet adhesion or MTA in unmodified blood samples compared to 40% HCT. This falls short of the in vitro and in vivo clotting enhancement reported by Walton et al. [19], in which 54% HCT resulted in larger thrombi and increased clotting speed. However, the similar SAC of thrombi formed by perfusion of unmodified 40 and 60% HCT is in agreement with the venous and arterial models presented by Lehmann et al. [21] and Chen et al. [50], respectively. The most notable difference between thrombi formed by 40 and 60% HCT blood was the difference in both SAC and MTA of A2A9-modified samples (Figs. 5, 6, 7). When αIIbβIII function was inhibited, 60% HCT blood did mediate increased platelet deposition relative to 40% HCT, indicating that GPIb-vWF adhesion was also increased due to elevated HCT. However, the loss of SIPA behavior in A2A9 blocked samples, as measured by both SAC and MTA, indicates that while GPIb-vWF bonds might dominate platelet adhesion at arterial shear rates, their ability to permit SIPA at supraphysiological shear is diminished greatly without subsequent stabilization by αIIbβIII-vWF bonds. This effect was independent of biochemically-induced platelet activation as blood citration was not reversed in this assay, preventing triggering of the coagulation cascade.
To provide further support for the reported trends, a control study was conducted in which PRP-glycerol mixtures were created to match the viscosities of 20, 40, and 60% HCT blood samples. This would ensure that increased platelet deposition was a result of platelet margination and not simply the result of increased shear stress relative to the 0% HCT case. Experiments conducted on these mixtures revealed no observable differences between platelet deposition of the PRP-glycerol or 0% HCT blood [Supplemental Fig. 5]. Thrombus formation was negligible for all conditions, demonstrating that the increase in viscosity corresponding to hematocrit levels was not responsible for the observed increase in thrombi formation reported here (Supplemental Table 1). While a positive correlation between elevated shear stress and increased platelet activation has been demonstrated thoroughly [64, 65], a lack of any increased deposition in this case indicates that elevated shear stress alone does not result in either increased platelet deposition or altered thrombus morphology. In blood experiments, we observed SIPA behavior in control samples at 40 and 60% HCT, but no such behavior when HCT (0 or 20%) was too low for effective platelet margination. Sufficient margination of platelets to the wall is a necessary prerequisite condition for increased platelet adhesion as a result of elevated shear stresses.
The depletion of ADP from ghost red blood cells allowed for the separation of biochemical and physical effects of red blood cells under supraphysiological shear rates [66]. Previous studies investigated shear-induced RBC secretions have reported complete loss of aggregation when ADP-consuming enzymes are used to prevent platelet-ADP signaling [67, 68]. Platelet adhesion, as measured by SAC, was entirely unaffected by reconstitution with ghost cells rather than native red blood cells (Supplemental Fig. 10). A lack of ADP secretion did not cause any reduction in overall platelet adhesion, further supporting that initial microthrombus formation at these shear rates is an entirely vWF-dependent process, in which platelet delivery to the vWF capture region is mediated by physical forces applied by red blood cells. Platelet aggregation (MTA) was mildly reduced in the presence of ghost cells, but this reduction was statistically significant at only one condition (Supplemental Fig. 11). MTA was most comparable at 5–10,000 s−1 between the 60% HCT unmodified and 60% HCT ghost cell conditions (Supplemental Fig. 12). Notably, SIPA was still observed as shear rate increased from 1000 to 10,000 s-1, and aggregation increased with increasing HCT. These data indicate that while ADP secretion might enhance SIPA, particularly when HCT is not elevated, physical forces on platelets are principally responsible for the aggregation reported, rather than biochemical signaling from red blood cells.
The inhibition of the GPIb receptor caused a dramatic drop in overall platelet adhesion (Supplemental Fig. 10) at 5000 and 10,000 s−1. SAC of GPIb-blocked and unmodified control samples was comparable at 1000 s-1, particularly at 60% HCT. This loss in adhesive capability was unsurprising, attachment of platelets at shear rates above this critical shear rate threshold of 1–1500 s−1 is dependent on fast-acting GPIb bonds that facilitate transient tethering prior to firm adhesion. Due to this lack of adhesion, MTA was similarly reduced drastically, as platelets did not permanently adhere to permit aggregation (Supplemental Fig. 11). The notable exception was the step region in Q3 (Supplemental Fig. 12); as local shear rates dropped below this critical shear rate threshold, deceleration permitted temporary platelet binding through GPIb-independent mechanisms (Supplemental Figs. 13–14). Following this, aggregation consistently occurred within this specific region, reaching thrombus sizes equal or greater to that of control samples. This behavior was observed at 5000 s-1 for both 40 and 60% HCT, and at 10,000 s−1 for 60% HCT. SIPA was consistent between samples at supraphysiological shear rates, as long as margination was sufficient and the local shear rate dropped below the critical shear rate threshold at which GPIb-dependent tethering occurs. GPIb-vWF binding is needed to permit any adhesion when the local shear rate exceeds this threshold, but aggregation will occur through αIIbβIII-vWF binding if the prerequisite initial platelet layer is allowed to form. On the contrary, while αIIbβIII permits limited platelet adhesion at all the investigated shear rates, SIPA is substantially prevented.
Additional validation tests were run post hoc, by normalizing SAC of individual donors with a correction factor based on the mean platelet concentration, and by separating male and female donors. Both of these assessments demonstrated that the trends observed had no dependence on bulk platelet concentration, or donor sex, despite reduced deposition in female donors relative to male.
There are limitations in this study. First, blood samples were not recalcified. This was done intentionally to elicit platelet-surface responses without inducing occlusive thrombus formation, but it would be physiologically relevant to replicate the study with an on-chip upstream mixing feature to recalcify blood samples and investigate the same effect. For experiments involving PRP-glycerol mixtures, while platelet adhesion was shown to not be increased by increasing shear stress alone, platelet activation was not assessed. As blood from male donors resulted in notably larger thrombi than those produced from the blood of female donors, it is possible that there were some differences in platelet activity or plasma protein levels between sexes that was not determined. Finally, plasma vWF levels were not measured in donors. While donors were screened for bleeding disorders, it would be informative to assess the effect of plasma vWF concentration of microthrombus variability.
Conclusions
This study investigated a number of parameters that contributed to platelet deposition within a sudden expansion channel. The effect of hematocrit on thrombus surface deposition and adhesive behaviors was investigated at arterial, pathological, and device-relevant shear rates. Within this sudden expansion framework, hematocrit, shear rate, and selective inhibition of the αIIbβIII receptor were observed to be the driving factors in determining the degree of platelet deposition.
-
1.
Reduction of hematocrit from physiological (40%) to 20% reduced platelet adhesion. Elevated (60%) hematocrit increased platelet adhesion relative to 40%. Formation of large aggregates through shear-induced platelet aggregation was dependent on 40% or 60% hematocrit at 5000 and 10,000 s−1.
-
2.
Increasing shear rate and hematocrit interacted to form larger thrombi as a result of overall binding behavior shifting to preferential platelet-platelet interactions rather than singular platelet-collagen interactions. This suggests the presence of a switch in platelet adhesion from collagen binding to aggregation that can be triggered by a combination of hematocrit and shear rate, a potentially useful finding in optimizing antiplatelet therapeutic agents for MCS patients. This shift in binding behavior is permitted by the αIIbβIII receptor.
-
3.
Platelet adhesion within this supraphysiological shear range is also tied strongly to integrin function. When αIIbβIII was inhibited, dramatic reductions in platelet deposition were observed. Elevated hematocrit still promoted modest platelet adhesion in opposition to αIIbβIII inhibition, particularly in the step region of the sudden expansion where flow deceleration occurred.
Data availability
Data will be made available by the corresponding author (K.B. Manning) upon reasonable request.
References
Sadler, J. E. Biochemistry and genetics of von Willebrand factor, 1998.
Siedlecki, C. A., B. J. Lestini, K. K. Kottke-Marchant, S. J. Eppell, D. L. Wilson, and R. E. Marchant. Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood. 88:2939–2950, 1996.
Ruggeri, Z. M., and J. Ware. von Willebrand factor. FASEB J. 7:308–316, 1993. https://doi.org/10.1096/fasebj.7.2.8440408.
Fu, H., Y. Jiang, D. Yang, F. Scheiflinger, W. P. Wong, and T. A. Springer. Flow-induced elongation of von Willebrand factor precedes tension-dependent activation. Nat Commun. 2017. https://doi.org/10.1038/s41467-017-00230-2.
Pareti, F. I., Y. Fujimura, J. A. Dent, L. Z. Holland, T. S. Zimmerman, and Z. M. Ruggeri. Isolation and characterization of a collagen binding domain in human von Willebrand factor. J Biol Chem. 261:15310–15315, 1986. https://doi.org/10.1016/S0021-9258(18)66869-3.
Houdijk, W. P. M., K. S. Sakariassen, P. F. E. M. Nievelstein, and J. J. Sixma. Role of Factor VIII-von Willebrand factor and fibronectin in the interaction of platelets in flowing blood with monomeric and fibrillar human collagen types I and III. J Clin Investig. 75:531–540, 1985. https://doi.org/10.1172/JCI111729.
Liu, Z. L., D. N. Ku, and C. K. Aidun. Mechanobiology of shear-induced platelet aggregation leading to occlusive arterial thrombosis: a multiscale in silico analysis. J Biomech. 2021. https://doi.org/10.1016/j.jbiomech.2021.110349.
Casa, L. D. C., D. N. Ku, and G. W. Woodruff. Thrombus formation at high shear rates. Annu Rev Biomed Eng. 2017. https://doi.org/10.1146/annurev-bioeng-071516.
Westein, E., A. D. Van Der Meer, M. J. E. Kuijpers, J. P. Frimat, A. Van Den Berg, and J. W. M. Heemskerk. Atherosclerotic geometries exacerbate pathological thrombus formation poststenosis in a von Willebrand factor-dependent manner. Proc Natl Acad Sci USA. 110:1357–1362, 2013. https://doi.org/10.1073/pnas.1209905110.
Manning, K. B., F. Nicoud, and S. M. Shea. Mathematical and computational modeling of device-induced thrombosis. Curr Opin Biomed Eng. 20:100349, 2021. https://doi.org/10.1016/J.COBME.2021.100349.
Kreuziger, L. B., M. S. Slaughter, K. Sundareswaran, and A. E. Mast. Clinical relevance of histopathologic analysis of heart mate II thrombi. ASAIO J. 64:754–759, 2018. https://doi.org/10.1097/MAT.0000000000000759.
Consolo, F., A. Dimasi, M. Rasponi, L. Valerio, F. Pappalardo, D. Bluestein, M. J. Slepian, G. B. Fiore, and A. Redaelli. Microfluidic approaches for the assessment of blood cell trauma: A focus on thrombotic risk in mechanical circulatory support devices. Int J Artif Organs. 39:184–193, 2016. https://doi.org/10.5301/ijao.5000485.
Kim, D. A., and D. N. Ku. Structure of shear-induced platelet aggregated clot formed in an in vitro arterial thrombosis model. Blood Adv. 6:2872–2883, 2022. https://doi.org/10.1182/bloodadvances.2021006248.
Colace, T. V., and S. L. Diamond. Direct observation of von Willebrand factor elongation and fiber formation on collagen during acute whole blood exposure to pathological flow. Arterioscler Thromb Vasc Biol. 33:105–113, 2013. https://doi.org/10.1161/ATVBAHA.112.300522.
Scavone, M., S. Bozzi, T. Mencarini, G. Podda, M. Cattaneo, and A. Redaelli. Platelet adhesion and thrombus formation in microchannels: the effect of assay-dependent variables. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21030750.
Receveur, N., D. Nechipurenko, Y. Knapp, A. Yakusheva, E. Maurer, C. V. Denis, F. Lanza, M. Panteleev, C. Gachet, and P. H. Mangin. Shear rate gradients promote a bi-phasic thrombus formation on weak adhesive proteins, such as fibrinogen in a von Willebrand factor-dependent manner. Haematologica. 105:2471–2483, 2020. https://doi.org/10.3324/haematol.2019.235754.
Rack, K., V. Huck, M. Hoore, D. A. Fedosov, S. W. Schneider, and G. Gompper. Margination and stretching of von Willebrand factor in the blood stream enable adhesion. Sci Rep. 2017. https://doi.org/10.1038/s41598-017-14346-4.
Zhao, R., M. V. Kameneva, and J. F. Antaki. Investigation of platelet margination phenomena at elevated shear stress. Biorheology. 44:161–177, 2007.
Walton, B. L., M. Lehmann, T. Skorczewski, L. A. Holle, J. D. Beckman, J. A. Cribb, M. J. Mooberry, A. R. Wufsus, B. C. Cooley, J. W. Homeister, R. Pawlinski, M. R. Falvo, N. S. Key, A. L. Fogelson, K. B. Neeves, and A. S. Wolberg. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood. 129:2537–2546, 2017. https://doi.org/10.1182/blood-2016-10-746479.
Tilles, A. W., and E. C. Eckstein. The near-wall excess of platelet-sized particles in blood flow: its dependence on hematocrit and wall shear rate. Microvasc Res. 33:211–223, 1987. https://doi.org/10.1016/0026-2862(87)90018-5.
Lehmann, M., R. M. Schoeman, P. J. Krohl, A. M. Wallbank, J. R. Samaniuk, M. Jandrot-Perrus, and K. B. Neeves. Platelets drive thrombus propagation in a hematocrit and glycoprotein VI–dependent manner in an in vitro venous thrombosis model. Arterioscler Thromb Vasc Biol. 38:1052–1062, 2018. https://doi.org/10.1161/ATVBAHA.118.310731.
Qi, Q. M., E. Dunne, I. Oglesby, I. Schoen, A. J. Ricco, D. Kenny, and E. S. G. Shaqfeh. In vitro measurement and modeling of platelet adhesion on VWF-coated surfaces in channel flow. Biophys J. 116:1136–1151, 2019. https://doi.org/10.1016/j.bpj.2019.01.040.
Wang, Y., M. Morabito, X. F. Zhang, E. Webb, A. Oztekin, and X. Cheng. Shear-induced extensional response behaviors of tethered von Willebrand factor. Biophys J. 116:2092–2102, 2019. https://doi.org/10.1016/j.bpj.2019.04.025.
Zhussupbekov, M., R. Méndez Rojano, W.-T. Wu, and J. F. Antaki. von Willebrand factor unfolding mediates platelet deposition in a model of high-shear thrombosis. Biophys J. 121:4033–4047, 2022. https://doi.org/10.1016/j.bpj.2022.09.040.
Sing, C. E., and A. Alexander-Katz. Elongational flow induces the unfolding of von willebrand factor at physiological flow rates. Biophys J. 2010. https://doi.org/10.1016/j.bpj.2010.01.032.
Goodman, P. D., E. T. Barlow, P. M. Crapo, S. F. Mohammad, and K. A. Solen. Computational model of device-induced thrombosis and thromboembolism. Ann Biomed Eng. 33:780–797, 2005. https://doi.org/10.1007/s10439-005-2951-z.
Yang, L., T. Neuberger, and K. B. Manning. In vitro real-time magnetic resonance imaging for quantification of thrombosis. Magn Reson Mater Phys Biol Med. 34:285–295, 2021. https://doi.org/10.1007/s10334-020-00872-2.
Taylor, J. O., R. S. Meyer, S. Deutsch, Keefe, and B. Manning. Development of a computational model for macroscopic predictions of device-induced thrombosis A. Biomech Model Mechanobiol. 15:1713–1731, 2016. https://doi.org/10.1007/s10237-016-0793-2.
Yang, L., N. Tobin, and K. B. Manning. Refining a numerical model for device-induced thrombosis and investigating the effects of non-Newtonian blood models. J Biomech. 120:110393, 2021. https://doi.org/10.1016/j.jbiomech.2021.110393.
Jamiolkowski, M. A., J. R. Woolley, M. V. Kameneva, J. F. Antaki, and W. R. Wagner. Real time visualization and characterization of platelet deposition under flow onto clinically relevant opaque surfaces. J Biomed Mater Res A. 103:1303–1311, 2015. https://doi.org/10.1002/jbm.a.35202.
Dimasi, A., M. Rasponi, J. Sheriff, W. C. Chiu, D. Bluestein, P. L. Tran, M. J. Slepian, and A. Redaelli. Microfluidic emulation of mechanical circulatory support device shear-mediated platelet activation. Biomed Microdevices. 17:1–11, 2015. https://doi.org/10.1007/s10544-015-0015-1.
Jamiolkowski, M. A., D. D. Pedersen, W.-T.T. Wu, J. F. Antaki, and W. R. Wagner. Visualization and analysis of biomaterial-centered thrombus formation within a defined crevice under flow. Biomaterials. 96:72–83, 2016. https://doi.org/10.1016/j.biomaterials.2016.04.022.
Brækkan, S. K., E. B. Mathiesen, I. Njølstad, T. Wilsgaard, and J. B. Hansen. Hematocrit and risk of venous thromboembolism in a general population. The Tromsø study. Haematologica. 95:270–275, 2010. https://doi.org/10.3324/haematol.2009.008417.
Fitzgibbon, S., A. P. Spann, Q. M. Qi, and E. S. G. Shaqfeh. In vitro measurement of particle margination in the microchannel flow: effect of varying hematocrit. Biophys J. 108:2601–2608, 2015. https://doi.org/10.1016/j.bpj.2015.04.013.
Liu, Z. L., C. Bresette, C. K. Aidun, and D. N. Ku. SIPA in 10 milliseconds: VWF tentacles agglomerate and capture platelets under high shear. Blood Adv. 6:2453–2465, 2022. https://doi.org/10.1182/bloodadvances.2021005692.
Ldc, C., and G. Se. Relative contributions of von Willebrand factor and platelets in high shear thrombosis. J Hematol Thromb Dis. 2016. https://doi.org/10.4172/2329-8790.1000249.
Kushchenko, Y. K., and A. V. Belyaev. Effects of hydrophobicity, tethering and size on flow-induced activation of von Willebrand factor multimers. J Theor Biol. 2020. https://doi.org/10.1016/j.jtbi.2019.110050.
Belyaev, A. V., and Y. K. Kushchenko. Biomechanical activation of blood platelets via adhesion to von Willebrand factor studied with mesoscopic simulations. Biomech Model Mechanobiol. 2023. https://doi.org/10.1007/s10237-022-01681-3.
Belyaev, A. V: Computer modelling of initial platelet adhesion during microvascular thrombosis. Russ J Numer Anal Math Model. 2019. https://doi.org/10.1515/rnam-2019-0020.
Bennett, J. S., J. A. Hoxie, S. F. Leitman, G. Vilaire, and D. B. Cines. Inhibition of fibrinogen binding to stimulated human platelets by monoclonal antibody. Proc Natl Acad Sci USA. 80:2417–2421, 1983. https://doi.org/10.1073/pnas.80.9.2417.
Bennett, J. S., S. J. Shattil, J. W. Power, and T. K. Gartner. Interaction of fibrinogen with its platelet receptor. Differential effects of α and γ chain fibrinogen peptides on the glycoprotein IIb-IIIa complex. J Biol Chem. 263:12948–12953, 1988. https://doi.org/10.1016/s0021-9258(18)37654-3.
HEPES-buffered saline. Cold Spring Harb Protoc. 2006, pdb.rec8786 (2006). https://doi.org/10.1101/pdb.rec8786
Milionis, A. S. I., Fragouli, D., Brandi, F., Athanassiou, A. Combination of lithography and coating methods for surface wetting control. In: Updates in Advanced Lithography. InTech (2013)
Hansen, R. R., A. A. Tipnis, T. C. White-Adams, J. A. Di Paola, and K. B. Neeves. Characterization of collagen thin films for von Willebrand factor binding and platelet adhesion. Langmuir. 27:13648–13658, 2011. https://doi.org/10.1021/la2023727.
Sakariassen, K. S., L. Orning, and V. T. Turitto. The impact of blood shear rate on arterial thrombus formation. Future Sci OA. 2015. https://doi.org/10.4155/fso.15.28.
Neeves, K. B., S. F. Maloney, K. P. Fong, A. A. Schmaier, M. L. Kahn, L. F. Brass, and S. L. Diamond. Microfluidic focal thrombosis model for measuring murine platelet deposition and stability: PAR4 signaling enhances shear-resistance of platelet aggregates. Journal of Thrombosis and Haemostasis. 6:2193–2201, 2008. https://doi.org/10.1111/j.1538-7836.2008.03188.x.
Restle, D. J., D. M. Zhang, G. Hung, J. L. Howard, F. Kallel, M. A. Acker, P. Atluri, and C. R. Bartoli. Preclinical models for translational investigations of left ventricular assist device-associated von Willebrand factor degradation. Artif Organs. 39:569–575, 2015. https://doi.org/10.1111/aor.12428.
Hariharan, P., M. Giarra, V. Reddy, S. W. Day, K. B. Manning, S. Deutsch, S. F. C. Stewart, M. R. Myers, M. R. Berman, G. W. Burgreen, E. G. Paterson, and R. A. Malinauskas. Multilaboratory particle image velocimetry analysis of the FDA benchmark nozzle model to support validation of computational fluid dynamics simulations. J Biomech Eng. 2011. https://doi.org/10.1115/1.4003440.
Spann, A. P., J. E. Campbell, S. R. Fitzgibbon, A. Rodriguez, A. P. Cap, L. H. Blackbourne, and E. S. G. Shaqfeh. The effect of hematocrit on platelet adhesion: experiments and simulations. Biophys J. 111:577–588, 2016. https://doi.org/10.1016/j.bpj.2016.06.024.
Chen, H., J. I. Angerer, M. Napoleone, A. J. Reininger, S. W. Schneider, A. Wixforth, M. F. Schneider, and A. Alexander-Katz. Hematocrit and flow rate regulate the adhesion of platelets to von Willebrand factor. Biomicrofluidics. 2013. https://doi.org/10.1063/1.4833975.
Vignoli, A., S. Gamba, P. E. J. van der Meijden, M. Marchetti, L. Russo, S. Tessarolo, C. Giaccherini, F. Swieringa, H. Ten Cate, G. Finazzi, J. W. M. Heemskerk, and A. Falanga. Increased platelet thrombus formation under flow conditions in whole blood from polycythaemia vera patients. Blood Transfus. 20:143–151, 2022. https://doi.org/10.2450/2021.0456-20.
Woolley, J. R., J. J. Teuteberg, C. A. Bermudez, J. K. Bhama, K. L. Lockard, R. L. Kormos, and W. R. Wagner. Temporal leukocyte numbers and granulocyte activation in pulsatile and rotary ventricular assist device patients. Artif Organs. 38:447–455, 2014. https://doi.org/10.1111/aor.12200.
Long, J. A., A. Ündar, K. B. Manning, and S. Deutsch. Viscoelasticity of pediatric blood and its implications for the testing of a pulsatile pediatric blood pump. ASAIO J. 51:563–566, 2005. https://doi.org/10.1097/01.mat.0000180353.12963.f2.
Denis, C., J. A. Williams, X. Lu, D. Meyer, and D. Baruch. Solid-phase von Willebrand factor contains a conformationally active RGD motif that mediates endothelial cell adhesion through the α(v)β3 receptor. Blood. 82:3622–3630, 1993. https://doi.org/10.1182/blood.v82.12.3622.3622.
Watt, K. W. K., T. Takagi, R. F. Doolittle, and T. Takagi. Amino acid sequence of the β chain of human fibrinogen. Biochemistry. 18:68–76, 1979. https://doi.org/10.1021/bi00568a011.
Zhao, R., J. N. Marhefka, F. Shu, S. J. Hund, M. V. Kameneva, and J. F. Antaki. Micro-flow visualization of red blood cell-enhanced platelet concentration at sudden expansion. Ann Biomed Eng. 36:1130–1141, 2008. https://doi.org/10.1007/s10439-008-9494-z.
Schneider, S. W., Nuschele, S., Wixforth, A., Gorzelanny, C., Alexander-Katz, A., Netz, R. R., Schneider, M. F. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers (2007)
Savage, B., Sixma, J. J., Ruggeri, Z. M.: Functional self-association of von Willebrand factor during platelet adhesion under flow (2002)
Nesbitt, W. S., E. Westein, F. J. Tovar-Lopez, E. Tolouei, A. Mitchell, J. Fu, J. Carberry, A. Fouras, and S. P. Jackson. A shear gradient-dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 15:665–673, 2009. https://doi.org/10.1038/nm.1955.
Rana, A., Westein, E., Niego, B., Hagemeyer, C. E. Shear-dependent platelet aggregation: mechanisms and therapeutic opportunities, 2019.
Fu, H., Jiang, Y., Wong, W. P., Springer, T. A. Single-molecule imaging of von Willebrand factor reveals tension-dependent self-association, 2021.
Dütting, S., M. Bender, and B. Nieswandt. Platelet GPVI: a target for antithrombotic therapy?! Trends Pharmacol Sci. 33:583–590, 2012. https://doi.org/10.1016/J.TIPS.2012.07.004.
Lin, J., Sorrells, M. G., Lam, W. A., Neeves, K. B. Physical forces regulating hemostasis and thrombosis: vessels, cells, and molecules in illustrated review, 2021.
Bozzi, S., Y. Roka-Moiia, T. Mencarini, F. Vercellino, I. Epifani, K. R. Ammann, F. Consolo, M. J. Slepian, and A. Redaelli. Characterization of the competing role of surface-contact and shear stress on platelet activation in the setting of blood contacting devices. Int J Artif Organs. 44:1013–1020, 2021. https://doi.org/10.1177/03913988211009909.
Ding, J., Z. Chen, S. Niu, J. Zhang, N. K. Mondal, B. P. Griffith, and Z. J. Wu. Quantification of shear-induced platelet activation: high shear stresses for short exposure time. Artif Organs. 39:576–583, 2015. https://doi.org/10.1111/aor.12438.
Reimers, R. C., S. P. Sutera, and J. H. Joist. Potentiation by red blood cells of shear-induced platelet aggregation: relative importance of chemical and physical mechanisms. Blood. 64:1200–1206, 1984.
Goldsmith, H., E. Kaufer, and F. McIntosh. Effect of hematocrit on adenosine diphosphate-induced aggregation of human platelets in tube flow. Biorheology. 32:537–552, 1995. https://doi.org/10.1016/0006-355X(95)00031-4.
Goldsmith, H. L., D. N. Bell, S. Braovac, A. Steinberg, and F. McIntosh. Physical and chemical effects of red cells in the shear-induced aggregation of human platelets. Biophys J. 69:1584–1595, 1995. https://doi.org/10.1016/S0006-3495(95)80031-7.
Acknowledgments
This work is supported, in part, by NIH HL136369.The project described was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1 TR002014. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We would like to thank Jessica C. Cardenas (University of Texas Health Science Center at Houston) for generously reading this manuscript.
Funding
This work is supported, in part, by NIH HL136369. The project described was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1 TR002014.
Author information
Authors and Affiliations
Contributions
K.B.M. and A.R. conceived and designed study; C.T.W., S.C.W., and S.A.R. performed experimental studies; C.T.W. analyzed data, interpreted experimental results, generated figures, and drafted manuscript; C.T.W., K.B.M. and A.R. edited and revised manuscript; K.B.M applied for and obtained funding; C.T.W., S.C.W., S.A.R., A.R., and K.B.M. approved final manuscript.
Corresponding author
Ethics declarations
Competing interest
K.B.M. has a financial interest in Cranial Devices, Inc, a company which could potentially benefit from the results of this research. The interest has been reviewed and is being managed by the University in accordance with its individual Conflict of Interest policy, for the purpose of maintaining the objectivity of research at The Pennsylvania State University. All other authors declare no conflicts.
Informed Consent
Informed consent was obtained from all human subjects in a Pennsylvania State University IRB approved protocol.
Additional information
Associate Editor Scott Simon oversaw the review of this article.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Watson, C.T., Ward, S.C., Rizzo, S.A. et al. Influence of Hematocrit Level and Integrin αIIbβIII Function on vWF-Mediated Platelet Adhesion and Shear-Induced Platelet Aggregation in a Sudden Expansion. Cel. Mol. Bioeng. 17, 49–65 (2024). https://doi.org/10.1007/s12195-024-00796-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12195-024-00796-0