
Abstract
Introduction
Tumor metastasis to the brain occurs in approximately 20% of all cancer cases and often occurs due to tumor cells crossing the blood-brain barrier (BBB). The brain microenvironment is comprised of a soft hyaluronic acid (HA)-rich extracellular matrix with an elastic modulus of 0.1–1 kPa, whose crosslinking is often altered in disease states.
Methods
To explore the effects of HA crosslinking on breast tumor cell migration, we developed a biomimetic model of the human brain endothelium, consisting of brain microvascular endothelial cell (HBMEC) monolayers on HA and gelatin (HA/gelatin) films with different degrees of crosslinking, as established by varying the concentration of the crosslinker Extralink.
Results and Discussion
Metastatic breast tumor cell migration speed, diffusion coefficient, spreading area, and aspect ratio increased with decreasing HA crosslinking, a mechanosensing trend that correlated with tumor cell actin organization but not CD44 expression. Meanwhile, breast tumor cell incorporation into endothelial monolayers was independent of HA crosslinking density, suggesting that alterations in HA crosslinking density affect tumor cells only after they exit the vasculature. Tumor cells appeared to exploit both the paracellular and transcellular routes of trans-endothelial migration. Quantitative phenotyping of HBMEC junctions via a novel Python software revealed a VEGF-dependent decrease in punctate VE-cadherin junctions and an increase in continuous and perpendicular junctions when HBMECs were treated with tumor cell-secreted factors.
Conclusions
Overall, our quantitative results suggest that a combination of biochemical and physical factors promote tumor cell migration through the BBB.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Brain metastasis occurs in approximately 20% of all cancer cases2,47 and remains a devastating prognosis due to limited treatment options. Evidence in the literature, including in vivo imaging, has shown that tumor cells are capable of metastasizing to the brain via the circulatory and lymphatic systems,2 and that metastasis occurring through the circulatory system requires the tumor cells to traverse the BBB in order to reach the brain parenchyma.6 However, the mechanisms governing how tumor cells cross the blood–brain barrier (BBB) are not well understood.
In one study, close physical contact with the abluminal surface of the blood vessel was crucial for the spread of tumor cells, which actively transmigrated through gaps in the wall of the vasculature but also engaged in vascular remodeling during extravasation.33 Unique to the BBB, tight junction proteins (e.g., claudins, occludins) are overexpressed, and act in conjunction with adherens junctions (e.g., vascular endothelial cadherin, VE-cadherin) to regulate barrier integrity and permeability.1 These junctional proteins link to the actin cytoskeleton through zonula occludens (e.g., ZO-1), which have been shown to regulate cell and junctional tension, cellular migration, barrier formation, and angiogenesis.65 Disruption of these junctions is linked with increased permeability of the BBB to cells and molecules and is implicated in several diseases,24 including cancer metastasis43 and glioblastoma, where microvascular leakiness correlates with histological tumor grade.55 Metastatic breast tumor cells reportedly secrete factors that promote increased tumor cell-BBB adhesion and disrupt or rearrange junctions, weakening the barrier and leading to tumor cell transmigration.8,37 Junction disruption can be considered a decrease of junctional proteins present at the cell boundary, while junction rearrangement is a change in the boundary presentation (e.g., phenotype or morphology) of the specific junctional protein. Junctional phenotype is thought to be linked with the stability and maturity of the cell–cell junction. For instance, a linear, continuous junction parallel to the cell boundary is reported as a stable junction, exhibited by cells with low tension.18,51 On the other hand, increased levels of cellular tension or contractility is linked with unstable, discontinuous junctions, which can take the form of punctate (e.g., dotted) or perpendicular (e.g., serrated) morphologies. Our recent development of a Python-based Junction Analyzer Program (JAnaP) allows us to quantitatively phenotype these cell–cell junctions in a healthy biomimetic BBB and in disease-associated states.25
Tumor cell-derived biochemical cues and physical interaction with brain endothelial cells can alter brain endothelial cell–cell junctions in such a way that it directs the mode of trans-endothelial migration. For instance, melanoma cells are reported to disrupt junctions, presumably through protease secretion, and induce endothelial cell apoptosis leading to paracellular transmigration.23 Other studies have shown that breast tumor cells can cross endothelial barriers utilizing both transcellular and paracellular pathways.22 Tumor cells also secrete endothelial-altering substances that can lead to an influx of calcium,56 glycocalyx degradation,22 and increased contractility,12,41 of the targeted endothelial cells, all of which are associated with enhanced tumor cell transmigration at cell–cell junctions.12,22,41,56 Furthermore, we and others have demonstrated that tumor cells can even physically displace endothelial cells and “incorporate” or “intercalate” into the endothelium,27,50,54 and we hypothesized that this process may also represent a distinct step in the extravasation of tumor cells through the brain endothelium. Hence, here we aimed to quantify how breast tumor cell biochemical factors and physical contact with the brain endothelium affect endothelial cell–cell junction organization, and ultimately, tumor cell incorporation into a biomimetic BBB microenvironment.
In addition to cell-secreted factors, cell functionality is profoundly influenced by its surrounding extracellular matrix (ECM).49 However, it is not yet well understood which aspects of the brain microenvironment play a role in cancer progression.21 Hyaluronic acid (HA) is a glycosaminoglycan that serves as a major building block of the brain ECM, which, unlike other parts of the body, is not highly organized and consists mainly of loosely crosslinked networks.45 HA in the brain is linked with small glycoproteins, as well as tenasins, the density of which plays a large role in the function of various neural processes. Thus, the disruption of these linkages45 as well as ECM rearrangement10 could play a large role in disease progression. Indeed, for other ECM models such as collagen gels, crosslinking (e.g., via lysyl oxidase) has been shown to increase ECM stiffness, making it more conducive to the progression of tumors and other diseases.4,14,31,40 While HA has important biological functions related to joint lubrication and wound healing, it also plays a role in the invasion of tumor cells, and, in the case of primary glioma tumors, HA is highly upregulated in the surrounding ECM.49
Because ECM structure (e.g., arrangement, stiffness, etc.) is known to play a significant role in healthy and diseased states of the brain, here we have investigated how the crosslinking of a brain-like ECM affects brain endothelial cells, tumor cells, and the interaction between the two cell types. We have used an HA/gelatin-based system with varying degrees of crosslinking to mimic the brain ECM and used live-cell microscopy and immunofluorescence imaging to quantify cellular migration, morphology, cell–cell junction presentation, and tumor cell incorporation into brain endothelial cell monolayers, thus providing insight into the interplay of ECM crosslinking and bi-cellular systems on breast cancer metastasis across the BBB.
Methods
Cell Culture
Human breast adenocarcinoma cells, MDA-MB-231s (American Type Culture Collection, Manassas, VA, USA), or the brain-seeking clones (MDA-MB-231-BR; generously provided by Dr. Toshiyuki Yoneda, Osaka University, Japan) were cultured in Dulbecco’s modified eagle’s medium (DMEM) with high glucose and l-glutamine supplemented with 1% penicillin/streptomycin 10,000 U/mL (Pen/Strep) and 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA) and used below passage 20 after purchase (parental cells) or acquisition (BR cells). Human brain microvascular endothelial cells, HBMECs (Cell Systems, Kirkland, WA, USA), were cultured under static conditions in flasks coated with 0.1% gelatin (VWR, Radnor, PA, USA) in RPMI-1640 supplemented with 20% FBS, 1% Pen/Strep, 2 mM l-glutamine (Thermo Fisher Scientific), 30 μg/mL endothelial cell growth supplement (ECGS) (Millipore Sigma), and 100 μg/mL heparin (Millipore Sigma) and used below passage 12. For VE-cadherin-GFP adenovirus amplification, 293A cells (Thermo Fisher Scientific) were cultured in medium composed of high glucose DMEM, 10% FBS, 0.1 mM MEM Non-Essential Amino Acids (NEAA) (Thermo Fisher Scientific), 2 mM l-glutamine, and 1% Pen/Strep. All cells were cultured in a climate-controlled incubator at 37 °C, with 5% CO2:95% air and 50% humidity. MDA-MB-231-BR cell authentication was confirmed via short tandem repeat (STR) testing (Laragen) after receiving them from Dr. Toshiyuki Yoneda and prior to carrying out experiments.
HA/Gelatin Film Formation
HA/gelatin films were formed using the HyStem-C kit (ESIBIO, Alameda, CA, USA). The kit contained four components: DG water, Glycosil (thiolated HA), Gelin (thiolated gelatin), and Extralink (thiol-reactive PEGDA crosslinker). Instructions from the kit were followed and were also described previously by Prestwich.53 Briefly, all components were thawed for 30–60 min. Glycosil, Gelin, and DG water were then briefly heated at 37 °C in a water bath to increase solubility. Glycosil and Gelin were then dissolved in 1 mL DG water, rocked for approximately 1 h at room temperature, then briefly re-heated in the water bath. Extralink was dissolved in DG water to 10%, then was further diluted into aliquots of 6, 4, and 1%. 24-well glass bottom plates (13 mm glass diameter) or 35 mm glass bottom dishes (14 mm glass diameter) (MatTek, Ashland, MA, USA) were plasma treated using a plasma cleaner with a 5 min pumping step to create a vacuum and a 2.5 min treatment on the “High” RF power setting (Harrick Plasma, Ithaca, NY, USA, PDC-001-HP (115 V)) to increase hydrophilicity. The plates were then treated with ultraviolet light for 5–10 min for sterilization. Gelin and Glycosil were mixed in a 1:1 ratio and then the Glycosil/Gelin solution was combined in a 4:1 ratio with the appropriate concentration of Extralink. The final concentration of Extralink within the films were 0.2, 0.8, 1.2, and 2%. Twenty microliters of solution were plated in each well, spread, and covered for gelation, the time of which varied by Extralink concentration (2%: 2–5 min, 1.2%: 3–6 min, 0.8%: 4–7 min, 0.2%: 6–9 min). Once crosslinked, medium was added, and the films were incubated at 37 °C for at least 30 min or until cells were ready to plate.
Tumor Cell Morphology and Migration Assays
HA/gelatin films were prepared as described above in 24-well glass bottom plates. For MDA-MB-231-BR cells, a 1:1 mixture of gelatin and HA without Extralink (0%) was also included. After soaking with medium in the incubator, 5 × 104 MDA-MB-231 or MDA-MB-231-BR cells were plated on top of the films and set-up to image as soon as the cells settled to the bottom of the plate. Images were collected via time-lapse phase-contrast microscopy and analyzed as described below in the data analysis section.
Tumor Cell Immunofluorescence Staining and Transfected HBMEC Fixation
For CD44 staining, 2 × 104 MDA-MB-231 cells were plated on HA/gelatin films formed with 0.2, 0.8, 1.2, and 2% Extralink in glass bottom dishes, or on dishes incubated with 20 μg/mL type I collagen for at least 1 h at 37 °C and washed three times with PBS. The next day, the cells were fixed in 3.7% formaldehyde (Millipore Sigma) for 10 min at room temperature, washed three times with PBS for 5 min each on a rocker, and permeabilized with 0.5% Triton-X 100 (Millipore Sigma) for 5 min. The samples were then washed again three times in PBS for 5 min each and blocked in 2% bovine serum albumin (BSA) (Millipore Sigma) for 1 h at room temperature. Cells were then incubated at 4 °C overnight with CD44 antibody (monoclonal CD44 antibody (156 3C11) Mouse mAb #3570, Cell Signaling Technologies, Danvers, MA, USA), which was dissolved in 2% BSA at a 1:100 ratio. Next, cells were rinsed with PBS and again blocked with 2% BSA for 1 h at room temperature and washed with PBS. Cells were then incubated with 1:500 Phalloidin-Alexa Fluor 488 (Thermo Fisher Scientific), 1:100 of secondary antibody (Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody Alexa Fluor 568, Thermo Fisher Scientific, A-11004), and 1:2500 of Hoechst 33342 (Thermo Fisher Scientific) diluted in PBS for 1 h at room temperature. HBMECs transfected with the VE-cadherin-GFP adenovirus and tumor cells stained with Cell Tracker (described below) were also fixed for 10 min in 3.7% formaldehyde, washed with PBS, and stained with 1:2500 of Hoechst 33342 for 1 h at room temperature. Finally, all samples were washed with PBS and stored at 4 °C until imaging via confocal microscopy.
Tumor-Conditioned Medium (TCM) Preparation
To prepare TCM, MDA-MB-231 cells were seeded at 25% confluency in a T-75 flask and cultured in HBMEC medium for 72 h. The medium was then collected, centrifuged at 300×g for 10 min and the supernatant was filtered through a 40 μm cell strainer to remove any debris. This post-strained solution was then mixed at a 1:1 ratio with control HBMEC medium and designated as TCM. The TCM-free conditions consisted of HBMEC medium that was spun down and strained to the same degree as that collected from the MDA-MB-231 cells and mixed at a 1:1 ratio with fresh HBMEC medium.
HBMEC Immunofluorescence Staining for Morphology and Junction Assays
HA/gelatin films were prepared as described above in 24-well glass bottom plates. After soaking in medium at 37 °C, 5 × 104 HBMECs were plated onto each film. Approximately 24 h later, the cells were treated with TCM or TCM-free (Control) medium and cultured for an additional 24 h. For the VEGF studies, HBMECs were treated with TCM containing 0.12 μg/mL VEGF monoclonal antibody (Thermo Fisher Scientific #26503) or mouse IgG2b isotype control (Thermo Fisher Scientific), or TCM-free medium 24 h after cell-seeding and cultured for an additional 24 h. For immunofluorescence staining, HBMECs were rinsed with warm PBS and fixed with 1% formaldehyde (Millipore Sigma) for 10 min at room temperature. After three subsequent 5-min washes with PBS, the fixed HBMECs were then permeabilized with 0.25% Triton-X 100 (Millipore Sigma) for 5 min at room temperature. The samples were then washed again with PBS three times for 5 min each, then blocked for 1 h at room temperature with 2% goat serum (Abcam, Cambridge, MA, USA). Primary antibodies (rabbit polyclonal IgG to ZO-1 antibody (H-300) and mouse monoclonal IgG to VE-cadherin antibody (F-8); Santa Cruz Biotechnology, Dallas, TX, USA, sc-10804 and sc-9989, respectively) were diluted 1:50 in 2% goat serum and added to the cells overnight at 4 °C. Samples were rinsed the next day with PBS, blocked again for 1 h at room temperature with 2% goat serum and then treated with secondary antibody (goat anti-rabbit Alexa Fluor 488, Abcam, ab150077; and goat anti-mouse Alexa Fluor 568, ThermoFisher Scientific) at a 1:100 dilution and 1:2500 Hoechst 33342 (ThermoFisher Scientific), in PBS, for 1 h at room temperature. Samples were then rinsed with PBS and fresh PBS was added for imaging via fluorescence microscopy and analyzed as described below in the data analysis section.
ELISA Assay for VEGF Detection
To quantify the concentration of VEGF in TCM and TCM-free conditions, a Human VEGF Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA) was used per the manufacturer’s instructions. The medium samples were collected as stated in the “TCM Preparation” method above and quantified immediately following medium preparation.
Fixed-Cell Epifluorescence Microscopy
Immunostained HBMECs were imaged on an Olympus IX83 inverted microscope using a × 60 oil objective and Olympus cellSens Software. Images were collected in a manner that maximized monolayer coverage within the respective image. HBMEC images were collected at the same exposure times for consistent analysis and have been enhanced via ImageJ, to the same extent for each channel, for improved visualization within this manuscript.
Adenovirus Amplification
The VE-cadherin-GFP adenovirus was a generous gift from Dr. William Luscinskas (Harvard Medical School), whose lab has previously described the construction of this vector.59 For amplification of VE-cadherin-GFP adenovirus, 293A cells cultured to ~ 80% confluency in 100 mm dishes. The 293A cells were then infected with the adenovirus and cultured until ~ 80–90% of all cells lifted off the dish. The cells were then collected and centrifuged at 1000 rpm. The resultant pellet was frozen at − 80 °C for 30 min and then thawed at 37 °C for 15 min. The freeze-thaw was repeated a total of three times. The lysed cells were centrifuged at 3000 rpm and the supernatant was saved and frozen at − 80 °C. This supernatant was then used to transfect HBMECs.
Tumor Cell Incorporation Assays
HA/gelatin films were prepared as described above in 24-well, glass bottom plates with 0.2, 0.8, 1.2, and 2% Extralink as well as a 1:1 mixture of gelatin and HA without Extralink (0%). 5 × 104–1 × 105 HBMECs were plated on the HA/gelatin films. For experiments fluorescently visualizing live VE-cadherin-GFP-expressing HBMECs’ cell–cell junctions, freshly plated and barely-attached HBMECs were transfected with 10 µL of the VE-cadherin-GFP adenovirus, as previously described by Hamilla et al.,27 and incubated for two days. To investigate the percent incorporation of metastatic breast tumor cells into HBMEC monolayers, live MDA-MB-231 or MDA-MB-231-BR cells were first stained with CellTracker Orange CMRA Dye (Thermo Fisher Scientific) per the manufacturer’s instructions. CellTracker Orange CMRA Dye is stated in product specifications to only transfer from parent cell to daughter cell and not in between neighboring cells. Because staining was carried out in the complete absence of HBMECs, transfer of dye between the two cell types in co-culture conditions is highly unlikely. Briefly, MDA-MB-231 or MDA-MB-231-BR cells were cultured in 6 well plates and equilibrated in HBMEC medium for 2 h. The cells were washed with PBS and then incubated with 2 mL of 0.5 µM CellTracker Orange dye in RPMI-1640 and 1% Pen/Strep for 15 min. Cells were then washed again with PBS and incubated for 30 min in full HBMEC medium. The stained cells were then trypsinized and 1 × 104–2.5 × 104 MDA-MB-231 or MDA-MB-231-BR cells were plated on top of HBMECs that had been cultured to monolayer on HA/gelatin films for two days prior to MDA-MB-231 or MDA-MB-231-BR cell seeding. These samples were imaged using time-lapse phase-contrast and fluorescence microscopy as described below.
Time-Lapse Microscopy
Live-cell phase contrast and fluorescence images were captured using an IX83 microscope (Olympus, Center Valley, PA, USA) with a × 10 objective (tumor cell migration assays on bare films), × 20 objective (tumor cell morphology assays on bare films, incorporation assays with non-transfected HBMECs and CellTracker-stained tumor cells), and × 60 objective (incorporation assays with VE-cadherin-GFP-expressing HBMECs and CellTracker-stained tumor cells). The Olympus cellSens Software (Olympus) was used to acquire the images. Phase contrast images of tumor cells on bare films were collected every 5 to 15 min for migration analysis, and additional images were captured at the end of the time-lapse on the second day for morphological analysis. The imaging regions for transmigration experiments were selected based on best monolayer coverage and the presence of MDA-MB-231 or MDA-MB-231-BR cells. The live-cell imaging chamber was maintained at 37 °C, 50% humidity, and 5% CO2:95% air surrounding the microscope stage.
Confocal Microscopy
A PerkinElmer confocal spinning disk microscope (PerkinElmer, Waltham, MA) was used to obtain 3D images of fixed cells using a × 40 water immersion objective. Vertical z-stacks were taken using appropriate filters using the Volocity 3D Image Analysis software. All acquisition settings were kept consistent between images within each experiment for CD44 imaging and slightly adjusted for best visualization for transmigration imaging. The ImageJ software (NIH; https://imagej.nih.gov) was used to reconstruct individual z-stack images into 3D, maximum-intensity and interpolated projections. Brightness of each channel in images of MDA-MB-231 cells stained for CD44 was adjusted separately but identically between each image to ensure that intensities could be compared. Brightness of each channel in images of MDA-MB-231 cells incorporating into HBMEC monolayers were adjusted separately for best visibility of appropriate parts of the cells; thus, intensities cannot be directly compared for these images.
Data Analysis
Tumor Cell Morphology and Migration
Tumor cell morphology was analyzed using ImageJ by manually tracing phase contrast images of live cells captured during or after time-lapse experiments in ImageJ. Cell circularity, solidity, and inverse aspect ratio were calculated as we have previously described.60 To track tumor cell migration on bare films, phase-contrast time-lapse images were acquired in 5-min intervals. The ImageJ Manual Tracking plugin was used to track the approximate centroid of each cell starting 5–6 h post-plating and analyzing for up to the next 8–9 h. Cells were not tracked if they went out of frame, divided during the tracking time, or were otherwise obstructed or hard to track. A custom Matlab (MathWorks, Natick, MA, USA) code was used to calculate cell speed, mean squared displacement, and diffusion coefficient of the migrating cells, as we have previously described in detail.60 Approximately 8 h (495 frames) of data were used for speed calculation, approximately 7 h (80–90 frames) were used for MSD calculation, and approximately 3.3 h (40 frames) were used for diffusion coefficient calculations. MDA-MB-231-BR cells were significantly clumped in all trials, and we note that cells in clumps were not chosen for migration analysis.
HBMEC Morphology and Junction Presentation
To quantitatively analyze HBMEC morphology and cell–cell junction presentation, we utilized our custom Python-based junction analyzer program (JAnaP) that we previously described.25 Please note that this program is available for download on GitHub (https://github.com/StrokaLab/JAnaP). Here, we integrated the ability to quantify an additional junction protein on the same cell. In addition to ZO-1, we co-stained the HBMECs for VE-cadherin. The waypoints generated on the ZO-1 image were projected to the VE-cadherin-tagged image, but the path between each waypoint was generated based on the intensities of the VE-cadherin image. This eliminated the need to re-waypoint (i.e., re-trace) cells for each immuno-stain. A threshold value of 15 was used to isolate ZO-1 junctions while a threshold of 5 was used to isolate VE-cadherin junctions. Continuous junctions are junction pieces coinciding with the cell edge for at least 15 consecutive pixels. Junctions coinciding with the cell edge for less than 15 pixels are categorized as perpendicular, if they have a relative aspect ratio greater than 1.2, or punctate if they have a relative aspect ratio less than 1.2. The relative aspect ratio is calculated by dividing the tip-to-tip distance (Tip Dist.), or maximum thickness, by the length of the junction coinciding with the cell edge, termed “path length” (Supplemental Fig. S1, adapted from25). In this manuscript, the percent of the cell edge expressing each junction presentation (calculated by dividing the cumulative sum of the path length of each junction type by the cell perimeter), as well as the tip-to-tip distance, are presented. The average tip-to-tip distance per cell for each junction type was calculated, providing a measure of the average maximum thickness of the continuous, perpendicular, and punctate junctions for each cell. The program-generated variables were calculated on a per cell basis, with all cells from every trial pooled together (n is approximately 200 cells). The percent difference, however, was calculated based on the average of each junction type for every trial (n = 3). The percent difference was calculated by taking the \(\left( {\% {\text{Junction}}_{\text{TCM}} - \% {\text{Junction}}_{\text{Control}} } \right) \cdot (\left( {\% {\text{Junction}}_{\text{TCM}} + \% {\text{Junction}}_{\text{Control}} } \right)/2)^{ - 1}\) for each junction type, for both ZO-1 and VE-cadherin.
Tumor Cell Incorporation
To quantify the time that tumor cells take to incorporate into HBMEC monolayers, and the percent of the tumor cells that incorporate into the monolayers, CellTracker-stained MDA-MB-231 or MDA-MB-231-BR cells were tracked on HMBEC monolayers by capturing images in 15-min intervals. First, the total number of tumor cells per frame was recorded at the start of each time-lapse sequence. Tracking was carried out for 40 frames (585 min). Cells that went out of frame before frame 40 were excluded from analysis, as well as cells deemed “untrackable” due to clumping, visual obstructions, or migration to areas of large gaps in the monolayer. An incorporating cell was subjectively identified based on the disappearance of a white halo surrounding the cell and transition to a 2D-flattened morphology in phase contrast images. If a cell was spread, but retained a halo and 3D appearance, it was counted as not incorporating and instead was likely spreading on top of the monolayer. Percent incorporation was calculated by dividing the total cells that incorporated into the monolayer by the total number of cells tracked in each frame. The start time for a cell to incorporate was marked as the first frame where the cell appeared to be spreading into the monolayer, or the very first frame if it was already spread. If cells incorporated and then exited the monolayer, only the first instance of incorporation was counted. If a cell divided before incorporating or before frame 40, it was counted as two separate cells. If cells divided after incorporation, it was counted as one cell. Cumulative percent incorporation was calculated by averaging the total percent of cells that had incorporated at each 15 min timepoint out of the total number of analyzable cells.
For migration analysis pre- and post-incorporation, select cells were manually traced in ImageJ. If cells remained completely rounded for 40 frames, they were tracked for 20 frames (285 min) and characterized as “non-incorporating.” Cells that incorporated were tracked for 6 frames (75 min) prior to spreading (i.e. “pre-incorporation”) and 6 frames (75 min) after they had fully incorporated (i.e. “post-incorporation”). If a cell started to incorporate within the first 6 frames (i.e., did not have a sufficient pre-incorporation phase to track) or rounded up again post-incorporation in less than 6 frames, the cell was omitted. Tumor cells that appeared to be spread-out but maintained a white halo, indicating a higher focal plane, and did not incorporate within 40 frames were interpreted as cells adhered to, and migrating on, the surface of the monolayer, and were entirely excluded from analysis. Three randomly selected HBMECs per frame were also traced for up to 8 frames (105 min) for three experiments (36–66 cells total). The average absolute value of the change in area or speed between each time point was calculated for a given cell by subtracting the area or speed at the previous time point from the area or speed at the current time point and taking the absolute value of the difference. These differences were then averaged for the time points analyzed for each cell.
Statistical Analysis
GraphPad Prism 8 (GraphPad, La Jolla, CA, USA) was used for all statistical analysis and graph preparation. Data was tested for normality using a D’Agostino-Pearson normality test, and some data within the same set did not follow a normal distribution. Thus, non-parametric tests were used for statistical analysis. If there were not sufficient values for a D’Agostino-Pearson test, a non-parametric test was used for consistency. If data was normally distributed in all comparison groups, a one-way analysis of variance (ANOVA) without assumed equal variances was used. For data that was not all normally distributed, a non-parametric Kruskal-Wallis ANOVA test with a Dunn’s multiple comparison post-hoc test was used and for data that was all normally distributed a Brown-Forsythe and Welch ANOVA with Games-Howell post-hoc test was used. All tests were carried out with p > 0.05 indicating not statistically different, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. A linear regression was used to compare the number of tumor cells at the start vs. monolayer quality, the percent difference in HBMEC junction presentation between TCM and control treatments, and the effect of Extralink concentration on junction tip-to-tip distance. A Mann–Whitney test was used to compare the TCM vs. TCM-free conditions for each junction type at each Extralink concentration. A t test was used to compare the VEGF concentration in TCM vs. TCM-free conditions. Errors bars represent standard error of the mean. All data represents pooled values from three independent trials unless otherwise noted in the figure captions.
Results
MDA-MB-231 and MDA-MB-231-BR Cells Become Smaller and Slower with Increased HA/Gelatin Crosslinking
ECM crosslinking has been shown to alter tumor cell phenotype and invasiveness, and HA is known to be one of the primary components of the brain’s ECM.4,14,40,45 Hence, we aimed to evaluate the migration and morphology response of MDA-MB-231 tumor cells to varying degrees of HA/gelatin film crosslinking by altering the concentration of Extralink, a PEGDA-based crosslinker, during film formation.68,75 We found that with increased Extralink concentration from 0.2 to 2%, MDA-MB-231 cells became significantly smaller (Figs. 1a and 1b) and more circular (Figs. 1a and 1c) and increased in inverse aspect ratio, circularity, and solidity (Figs. 1a and 1c). Furthermore, MDA-MB-231 cells plated on HA/gelatin films with increased Extralink concentrations demonstrated more homogeneous morphologies as evidenced by a decreased spread of the data with higher Extralink concentrations (Figs. 1b and 1c). Additionally, MDA-MB-231 cell speed decreased with increased crosslinking of HA/gelatin films (Fig. 1d). Plots of mean squared displacement vs. time indicated that MDA-MB-231 cells explored smaller areas with increased crosslinking of HA/gelatin films (Fig. 1e), with a non-statistically significant decrease in diffusion coefficient with increased crosslinking of HA/gelatin films (Fig. 1f). Because a mesenchymal, metastatic phenotype is typically associated with highly migratory, elongated cells,58,70 these changes in cell morphology and migration could be relevant to the cells’ functionality.

MDA-MB-231 morphology and migration parameters on HA/gelatin films with varying degrees of crosslinking. (a) Phase contrast images of MDA-MB-231 cells on HA/gelatin films with 0.2, 0.8, 1.2, and 2% Extralink. Scale bar is 50 μm and applies to all images. (b) Area, (c) inverse aspect ratio, circularity, and solidity, (d) speed, (e) mean squared displacement, and (f) diffusion coefficient of MDA-MB-231 cells on HA/gelatin films with 0.2, 0.8, 1.2, and 2% Extralink. Means of columns that do not share a lower-case letter are significantly different with p < 0.05 via a non-parametric Kruskal–Wallis ANOVA test with a Dunn’s multiple comparison post-hoc test. (b, c): 529 ≤ N ≤ 582, (d): 142 ≤ N ≤ 228 where N is the number of cells. (e, f): N = 3, where N is the number of trials. All error bars represent standard error of the mean. All values are pooled from three independent trials.
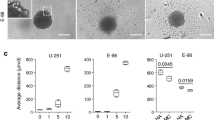
Due to the non-random metastasis of breast tumor cells to the brain,13,29 we also investigated the morphology and migration of MDA-MB-231-BR cells, the brain-seeking clone of the parental MDA-MB-231 cells.73 Prior work has shown distinct proteomic differences between brain-seeking and parental MDA-MB-231 cells in pathways related to cell survival, proliferation, movement, cell cycle, and signaling, suggesting that brain-seeking cell interaction with the BBB could differ from those of parental cells.19 When MDA-MB-231-BR cells were seeded on bare HA/gelatin films, phase contrast images revealed modest morphological differences between single cells on HA/gelatin films with different degrees of crosslinking (Fig. 2a). MDA-MB-231-BR cells in general formed more clusters than their parental counterparts (i.e., compared to Fig. 1a), and this clustering appeared to increase with increased HA/gelatin crosslinking (Fig. 2a). For subsequent analysis, we chose to focus on single MDA-MB-231-BR cells, not cells within the clusters, in order to keep analysis methods consistent between the two cell types. MDA-MB-231-BR single cell area decreased with increasing HA/gelatin crosslinking from 0 to 2%, 0.2 to 1.2%, 0.2 and 2% (Fig. 2b). There were no significant differences in inverse aspect ratio, circularity, or solidity of the single MDA-MB-231-BR cells on HA/gelatin films with different degrees of crosslinking (Fig. 2c), which was in contrast to the increase in these parameters for the parental cells (Fig. 1c). These results are likely due to the rounder MDA-MB-231-BR cells forming clusters on HA/gelatin films with higher crosslinking, leaving the larger, more elongated MDA-MB-231-BR cells as single cells.

MDA-MB-231-BR morphology and migration parameters on HA/gelatin films with varying degrees of crosslinking. (a) Phase contrast images of MDA-MB-231-BR cells on HA/gelatin films with 0, 0.2, 0.8, 1.2, and 2% Extralink. Scale bar is 50 μm and applies to all images. (b) Area, (c) inverse aspect ratio, circularity, and solidity, (d) speed, (e) mean squared displacement, and (f) diffusion coefficient of MDA-MB-231-BR cells on HA/gelatin films with 0, 0.2, 0.8, 1.2, and 2% Extralink. A non-parametric Kruskal–Wallis ANOVA with a Dunn’s multiple comparisons test was used for statistical analysis (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05). (b–d): N = 60 where N is the number of cells. (e, f): N = 3, where N is the number of trials. All error bars represent standard error of the mean. All values are pooled from three independent trials.
The migration speed of single MDA-MB-231-BR cells also decreased monotonically with increased HA/gelatin crosslinking density (Fig. 2d), similar to the trend for parental MDA-MB-231 cells (Fig. 1d). The mean squared displacements (Fig. 2e) and diffusion coefficients (Fig. 2f) were quantitatively more similar for MDA-MB-231-BR cells across HA/gelatin crosslinking densities in comparison with MDA-MB-231 parental cells (Figs. 1e and 1f).
Cell attachment to HA is mediated by CD44, which has also been implicated in transducing HA stiffness cues and associated with brain tumor progression and invasion.11,34,49,74 For breast tumor cells, CD44 increases tumor cell adhesion to and invasion of the endothelium, increasing the efficiency of distant metastasis.44 Hence, we explored whether the morphological and migratory behavior with altered HA/gelatin film crosslinking were also associated with differences in CD44 binding. Interestingly, MDA-MB-231 cells immunostained for CD44 on HA/gelatin films with varying degrees of crosslinking, as well as type I collagen-coated glass, did not present observable differences in CD44 expression (Fig. 3).

CD44 and actin staining in MDA-MB-231 parental cells. Reconstructed confocal z-stacks of MDA-MB-231 cells immunostained for CD44 (red) and stained for actin (green) on HA/gelatin films with 0.2, 0.8, 1.2, and 2% Extralink, as well as on glass coated with type I collagen. Scale bar on first image is 50 μm and applies to all images. Scale bar in the zoomed in inserts is 25 μm. Green: actin; red: CD44, blue: DNA. All images were reconstructed from a z-stack in ImageJ and intensities were adjusted equally for each channel and image.
Meanwhile, actin arrangement was altered, with MDA-MB-231 cells on type I collagen and HA/gelatin films with 0.2% Extralink displaying a somewhat fibrous actin arrangement (Fig. 3) which became more diffuse with increased film crosslinking, especially on the films with 2% Extralink (Fig. 3). Increased cell spreading and mesenchymal migration are typically correlated with a more stress fiber-rich actin arrangement,67 which is indeed in line with our observations. A stress fiber-rich actin cytoskeleton is also often linked with a high degree of cellular contractility, which also correlates with the stiffness of the underlying cellular matrix.17 Since increased crosslinking typically results in increased material stiffness, changes in film stiffness could drive alterations in tumor cell contractility and actin filament arrangement. Hence, we used atomic force microscopy to probe the effect of Extralink concentration on the Young’s modulus of HA/gelatin films (Supplemental Method S1). The average modulus of the films formed with 0.2, 0.8, 1.2, and 2% Extralink were measured to be 0.85, 1.1, 1.5, and 3.8 kPa, respectively (Supplemental Fig. S2). Interestingly, only the Young’s modulus of the film formed using 2% Extralink was statistically different from the Young’s modulus of the other film compositions. Despite this, we still found significant differences in MDA-MB-231 cell phenotype and behavior on those film compositions (Figs. 1 and 2).
HA/Gelatin Crosslinking Does Not Affect HBMECs Morphology or Junction Presentation
Metastasis across the BBB requires that tumor cells not only overcome the endothelial cell barrier, but also the underlying basement membrane. Tumor cells are reported to actively degrade the basement membrane by secreting matrix metalloproteinases (MMPs).69 Disruption of the basement membrane could lead to direct endothelial cell interaction with the brain ECM, and possibly abluminal HA-binding. Given the significant differences in MDA-MB-231 behavior on HA/gelatin films of varying crosslink density, we next wanted to understand how these differences in film composition might affect HBMEC monolayers. After two days of static culture on each film composition, HBMECs were fixed and immunostained for ZO-1 and VE-cadherin (Fig. 4a). In general, the percent of Extralink crosslinker did not affect the area (Fig. 4b), solidity (Fig. 4c), circularity (Fig. 4c), or perimeter (Supplemental Fig. S3) of individual HBMECs within a monolayer. To assess how HA/gelatin film crosslinking affects cell–cell junctions, we used the JAnaP25 to quantify the percent of the cell perimeters presenting different junction types for ZO-1 and VE-cadherin (Figs. 4d and 4e). HBMECs on HA/gelatin films with 0.8% Extralink presented the highest amounts of continuous, punctate, and perpendicular ZO-1 (Fig. 4d). For VE-cadherin, no difference in continuous or perpendicular junction presentation was observed across the different film compositions, but punctate junctions were highest on HA/gelatin films with 0.8 and 2.0% Extralink (Fig. 4e). We note that these results, and the results described below, were obtained for our cell culture models under static conditions and in the absence of other BBB cells such as astrocytes and pericytes; we discuss these limitations further in the Discussion section below.

HBMEC morphology and junction presentation on HA/gelatin films with varying degrees of crosslinking. (a) Immunofluorescence images of HBMECs immunostained for ZO-1 (green) and VE-cadherin (red) on HA/gelatin films with 0.2, 0.8, 1.2, and 2% Extralink. Scale bar on bottom right image is 20 μm and applies to all images in this panel. (b) Area and (c) solidity and circularity on HA/gelatin films with varying Extralink. The percent of the cell edge presenting continuous, punctate, and perpendicular junction for (d) ZO-1 and (e) VE-cadherin. The inset graphs depict the same perpendicular results, zoomed in to improve visibility. (b–e): 182 ≤ N ≤ 223, where N is the number of cells. The percent of the cell expressing continuous, punctate, and perpendicular junction is presented in (d) for ZO-1 and (e) for VE-cadherin. A non-parametric Kruskal–Wallis ANOVA with a Dunn’s multiple comparisons test was used for statistical analysis to compare results within each morphological parameter or junction type (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05). All error bars represent standard error of the mean and dots for panels b–e represent values for individual cells.
Tumor Cell Conditioned Media Alters HBMEC Junction Presentation
To understand the effects of biochemical signaling between HBMECs and metastatic tumor cells, and how these signaling effects may be modulated by HA/gelatin matrix crosslinking, we treated HBMECs with TCM. TCM-treated HBMECs presented visually thick, continuously connected junctions with a largely jagged morphology (Fig. 5a). The cells were smallest on HA/gelatin films made with 2.0% Extralink (Fig. 5b, Supplemental Fig. S3), and were significantly smaller than HBMECs not treated with TCM (Fig. 4b) on 0.8 and 2.0% Extralink. However, no changes in circularity or solidity were observed between Extralink concentrations or due to TCM treatment (Figs. 4c and 5c). While no apparent trend was observed for VE-cadherin junctions on varying Extralink films in response to TCM, ZO-1 junctions generally decreased with increased Extralink, except continuous junctions on 0.2% (Figs. 5d and 5e).

TCM-treated HBMEC morphology and junction presentation on HA/gelatin films with varying degrees of crosslinking. (a) Immunofluorescence images of HBMECs treated with TCM on HA/gelatin films with 0.2, 0.8, 1.2, and 2% Extralink. Scale bar on bottom right image is 20 μm and applies to all images in this panel. (b) Area and (c) solidity and circularity on HA/gelatin films with varying Extralink. The percent of the cell edge presenting continuous, punctate, and perpendicular junction for (d) ZO-1 and (e) VE-cadherin. The inset graphs depict the same perpendicular junction results but zoomed in to improve visibility. (b–e): 195 ≤ N ≤ 224, where N is the number of cells. The percent difference of (f) ZO-1 and (g) VE-cadherin between TCM (d, e) and TCM-free (Figs. 4d and 4e) for each junction type. (f, g): N = 3, where N is the number of trials. Legend in panel G applies to panels (f) and (g). The colored dotted lines represent the linear regression (Lin. Reg.) of each respective junction type. The tip-to-tip distance (TipDist.) of (h) ZO-1 and (i) VE-cadherin, where the dotted and solid lines represent the linear regression of the TCM-free and TCM conditions, respectively. (h, i): 86 ≤ N ≤ 129, where N is the number of cells. Legend in panel (i) applies to panels (h) and (i). A non-parametric Kruskal–Wallis ANOVA with a Dunn’s multiple comparisons test was used for statistical analysis to compare results within each morphological parameter or junction type (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05). For (f, g), statistics represent the difference between TCM and TCM-free conditions presented in (d) and (e) and Figs. 4d and 4e, respectively, calculated using a Mann–Whitney test for each condition. For (h, i), statistics represent the difference between TCM vs. TCM-free results for each junction type at the respective Extralink concentration within the respective graph. All error bars represent standard error of the mean and dots for panels (b–e) represent values for individual cells. Linear regression statistical analysis rendered all fits non-significantly non-zero.
To investigate the specific effects of TCM treatment on HBMEC junctional protein presentation, we calculated the percent difference between the TCM (Figs. 5d and 5e) and TCM-free conditions (Figs. 4d and 4e) for each HBMEC junction type and film composition. For ZO-1, TCM treatment increased continuous junction presentation on all films except on the film with the highest Extralink concentration. A linear fit (slope = − 33.0% difference/% Extralink, R2 = 0.769) suggests a decreasing trend with increasing Extralink concentration, though the slope was not significantly non-zero (Fig. 5f). While perpendicular junctions responded with a similar trend (slope = − 31.7% difference/% Extralink, R2 = 0.794, non-significant non-zero slope), punctate ZO-1 was much less sensitive to TCM (slope = − 7.2% difference/% Extralink, R2 = 0.269, non-significant non-zero slope) (Fig. 5f). For VE-cadherin, both continuous and perpendicular junctions increased with TCM for all film compositions, while punctate junctions generally decreased. The response of VE-cadherin junction presentation to TCM was even less affected by the Extralink concentration compared to ZO-1, as the slope of the continuous, perpendicular, and punctate linear regressions were − 3.1, 6.7, and − 2.8% difference/% Extralink, respectively, and all were non-significantly non-zero (Fig. 5g).
Though quantification of junction coverage provides insight into the localization of each junctional protein, it does not provide an explicit measure of junction thickness or “protrusion”. We therefore analyzed the average tip-to-tip distance of each junction type to quantify the maximum distance the junction protruded from the cell edge for ZO-1 (Fig. 5h) and VE-cadherin (Fig. 5i). As expected, the punctate junctions (presented in red in Figs. 5h and 5i) had the smallest tip-to-tip distance compared to continuous and perpendicular junctions. For ZO-1 (Fig. 5h), the perpendicular junctions had the largest tip-to-tip distance. This is in line with our expectations, since by definition, perpendicular junctions protrude radially outward from the cell edge.18 Minimal change in ZO-1 tip distance was observed, except for punctate junctions which increased with TCM on all films except the 2.0% Extralink film. On the other hand, TCM increased the protrusion thickness of VE-cadherin junctions in nearly every condition. Interestingly, the continuous and perpendicular junctions were nearly the same width, suggesting that the continuous junctions did not take the form of linear, mature junctions parallel to the cell border,51 rather protrusive perpendicular-like junctions that continuously presented themselves around the cell edge. Notably, the tip-to-tip distance of VE-cadherin was always greater than ZO-1 for each respective junction type. These results were in line with previous studies that reported manual width measurements of tight junctions (specifically, claudin-5) to be approximately 1 to 1.5 μm wide32 and VE-cadherin junctions to be approximately 1 to 3 μm wide.35 Overall, Extralink concentration did not affect the tip-to-tip distance results, since all the linear regression fits returned non-significantly non-zero slopes, though some significant differences did emerge between groups (Supplemental Fig. S4).
Metastatic breast tumor cells secrete Vascular Endothelial Growth Factor (VEGF) which can disrupt or rearrange cell–cell junctions,8,37 or induce elevated expression of Angiopoietin-2 by endothelial cells, further weakening the monolayer.8 To probe whether VEGF was playing a role in the junctional changes observed with TCM treatment, we first checked to see if VEGF was in fact present in the TCM produced by MDA-MB-231 cells. Using an ELISA, we found that VEGF was present in TCM at approximately 9000 pg/mL, a concentration significantly greater than the TCM-free control medium (approximately 3 pg/mL) (Fig. 6a). We therefore investigated the effects of inhibiting VEGF in TCM on junction presentation. To do this, we seeded HBMECs on HA/gelatin films composed of 0.8% Extralink. As expected, the HBMECs treated with the TCM control presented thick, jagged junctions (Fig. 6b), similar to those observed with TCM treatment (Fig. 5a). When VEGF was blocked, however, the junctions lost this perpendicular phenotype, and instead presented junctions resembling the TCM-free condition (Fig. 6b). Junction quantification showed a significant decrease in perpendicular junction coverage and thickness (Tip Dist.) for both ZO-1 (Figs. 6c and 6e) and VE-cadherin (Figs. 6d and 6f) with VEGF inhibition, but little to no significant difference between the anti-VEGF and TCM-free conditions. Furthermore, VEGF inhibition not only decreased continuous VE-cadherin coverage, but it also decreased the thickness of both continuous ZO-1 (Fig. 6e) and continuous VE-cadherin (Fig. 6f), verifying a shift from jagged junctions to the more linear phenotype observed in the absence of TCM. Overall, these studies implicate tumor cell-secreted VEGF in the observed alterations in junctional appearance with TCM treatment.

Role of tumor cell-secreted VEGF in HBMEC junction presentation. (a) ELISA quantification of VEGF in TCM vs. TCM-free control (N = 3 biological replicates). (b) Immunofluorescence images of HBMECs treated with TCM Control (TCM + isotype antibody), anti-VEGF (TCM + VEGF antibody), and TCM-Free control on 0.8% Extralink film. Scale bar on bottom right image is 20 μm and applies to all images in this panel. The percent of the cell edge presenting continuous, punctate, and perpendicular junctions for (c) ZO-1 and (d) VE-cadherin. The tip-to-tip distance (Tip Dist) of (e) ZO-1 and (f) VE-cadherin. (c, d): 77 ≤ N ≤ 82 and (e, f): 71 ≤ N ≤ 82, where N is the number of cells. A t test was used for statistical comparison for panel (a), while a non-parametric Kruskal–Wallis ANOVA with a Dunn’s multiple comparisons test was used for panels (c–f) to compare results within each junction type (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, NS p > 0.05). All error bars represent standard error of the mean and dots for panels (c–f) represent values for individual cells.
MDA-MB-231 and MDA-MB-231-BR Cells Incorporate into HBMEC Monolayers
Because we observed that tumor cell-derived biochemical cues from TCM can alter HBMEC junction morphology, we next examined the effect of HA/gelatin film crosslinking on breast tumor cell incorporation into HBMEC monolayers. Phase contrast and fluorescence microscopy were used to evaluate CellTracker-stained MDA-MB-231 or MDA-MB-231-BR incorporation into HBMEC monolayers (Fig. 7a). We found no statistically significant difference in the percent incorporation of MDA-MB-231 cells into HBMECs (Fig. 7b) or in the time from the start of the time-lapse to complete incorporation (Fig. 7c) as a function of Extralink concentration under static conditions. This was not surprising, since the alterations in HBMEC cell–cell junction presentation due to TCM-treatment were mostly not affected by the Extralink concentration of the underlying HA/gelatin films (Figs. 5f–5i). The cumulative percent of MDA-MB-231 cell incorporation, or percent of incorporation at each time point, was generally similar for all films, regardless of Extralink concentration under static conditions (Fig. 7d). Furthermore, we found that the plot of percent MDA-MB-231 cell incorporation at ~ 10 h vs. the average number of MDA-MB-231 cells at the start of the time-lapse had a slope that was not significantly non-zero (Fig. 7e).

MDA-MB-231 and MDA-MB-231-BR cell incorporation into HBMEC monolayers on HA/gelatin films with varying degrees of crosslinking. (a) Phase contrast images of an incorporating (blue arrow) and a non-incorporating (green arrow) CellTracker-stained MDA-MB-231 cell (red) on an HBMEC monolayer. (b) Percent of MDA-MB-231 cells that incorporate (12 ≤ N ≤ 22, where N is the number of time-lapse sequences analyzed per condition), (c) time from start of time-lapse to full incorporation of MDA-MB-231 cells, and (d) the cumulative percent incorporation of MDA-MB-231 cells over time (65 ≤ N ≤ 184, where N is the number of incorporating cells) into HBMEC monolayers on HA/gelatin films with 0, 0.2, 0.8, 1.2, and 2% Extralink in a total time of 9.75 h. (e) Average percent incorporation as a function of the number of MDA-MB-231 cells in frame at the start of the time-lapse. (f) Percent of MDA-MB-231-BR cells that incorporate (N = 6, where N is the number of time-lapse sequences analyzed per condition), (g) time from start of time-lapse to full incorporation of MDA-MB-231-BR cells, and (h) the cumulative percent incorporation of MDA-MB-231-BR cells over time (45 ≤ N ≤ 62 where N is the number of incorporating cells) into HBMEC monolayers on HA/gelatin films with 0, 0.2, 0.8, 1.2, and 2% Extralink in a total time of 9.75 h. For panels B, C, F, p > 0.05 via a non-parametric Kruskal–Wallis ANOVA test with a Dunn’s multiple comparison post-hoc test between all groups. For panel G, a Brown–Forsythe and Welch ANOVA with a Games–Howell multiple comparison post-hoc test was used with*p ≤ 0.05, **p ≤ 0.01. All error bars represent standard error of the mean. All data is pooled from three independent trials.
Interestingly, we observed similar results for the MDA-MB-231-BR cells, with some modest differences. Similar to MDA-MB-231 cells, there was no difference in the percent incorporation of MDA-MB-231-BR cells as a function of HA/gelatin crosslinking density (Fig. 7f). Meanwhile, MDA-MB-231-BR cells took longer to complete incorporation (starting from time of plating) on the HA/gelatin films with 0% Extralink in comparison with 0.2, 0.8, and 1.2% conditions (Fig. 7g). The cumulative percent of MDA-MB-231-BR cell incorporation, or percent of incorporation at each time point, was generally similar for all films, regardless of Extralink concentration under static conditions (Fig. 7h). Notably, the average percent incorporation was generally lower for MDA-MB-231-BR cells as compared to parental cells, with 43–52% of MDA-MB-231 parental cells incorporating (Fig. 7b), and 37–47% of MDA-MB-231-BR cells incorporating during the first approximately 10 h post-plating (Fig. 7f). The magnitudes of the time from tumor cell plating to complete incorporation remained relatively similar (Figs. 7c and 7g).
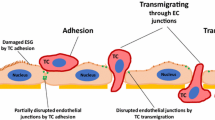
Tumor cells can utilize multiple modes of extravasation across endothelial cell barriers, and hence we used confocal and live-cell microscopy to image CellTracker-stained MDA-MB-231 cells interacting with VE-cadherin-GFP-expressing HBMECs. We found that some MDA-MB-231 cells remained on top of or found their way underneath the HBMECs (Fig. 8a). As expected, many tumor cells simply squeezed paracellularly into gaps between HBMECs, with protrusions going above and/or underneath the monolayer (Figs. 8b and 8c (left insert)). We also found several interesting and unexpected interactions. In some cases, an HMBEC, identified by its VE-cadherin-GFP expression, contained an extra nucleus and internal CellTracker stain that should have exclusively stained the tumor cells (Figs. 8c (right insert), 8e, and 8f). We note that CellTracker Orange CMRA is transformed into a cell-impermeant dye once inside the cell and is activated via enzymatic cleavage and thus can spread to daughter cells but not neighboring cells, and thus we do not expect HBMECs to be nonspecifically labeled with CellTracker. By observing the 3D and cross-sectional views as well as the orthogonal view of the cells, it appeared that a tumor cell had transcellularly incorporated into the HBMEC monolayer, becoming a part of the HBMEC (Figs. 8c (right insert), 8e, and 8f). A different mode of tumor cell-HBMEC interaction was observed in HBMECs that appeared to contain CellTracker stain but retained solely one nucleus (Fig. 8d). The punctate nature of the CellTracker stain in these HBMEC leads us to speculate that there could be cellular material shed by tumor cells and internalized by HBMECs (Fig. 8d).

Confocal images of various modes of MDA-MB-231 cell incorporation into HBMEC monolayers. Confocal z-stacks reconstructed in ImageJ into a 3D stack showing evidence of (a) MDA-MB-231 cells on top of or underneath HBMECs, (b) paracellular incorporation of MDA-MB-231 with long cell tail above HBMEC monolayer, (c) paracellular and potentially transcellular incorporation, (d) potentially transcellular incorporation or spread of MDA-MB-231 cell material into HBMEC, (e) and (f) potentially transcellular incorporation of MDA-MB-231 cells into HBMEC monolayer. Intensity of each channel in each image was separately adjusted for better visualization; thus, relative intensities should not be compared. Scale bars from bottom row (panel f) apply to all images above except for the zoomed images. Each zoomed image is scaled individually, and all scale bars on zoomed images are 50 μm. All images were taken on HA/gelatin films with 0% Extralink. Green: VECadGFP (HBMEC); red: CellTracker (MDA-MB-231), blue: DNA (HBMEC and MDA-MB-231).
MDA-MB-231 Tumor Cells Dynamically Interact with HBMEC Junctions
To quantitatively capture the dynamics of tumor cell incorporation into HBMECs, we collected time-lapse images of GFP-VE-cadherin-expressing HBMECs and CellTracker-stained MDA-MB-231 tumor cells. We found that some MDA-MB-231 cells were able to migrate on top of HBMEC adherens junctions and appeared to morphologically “unzip” them (Fig. 9a, white arrow), such that the adherens junctions opened and then closed behind the tumor cell (Fig. 9a, yellow arrow). Additionally, we found that MDA-MB-231 cells could squeeze in between HBMECs, resulting in complete disruption of the surrounding adherens junctions (Fig. 9b, magenta and purple arrows). Similar to Figure 8d, some image sequences suggested that HBMECs internalized cellular material shed by MDA-MB-231 cells, without visually affecting surrounding adherens junctions (Fig. 9b, blue arrow). These time-lapse sequences allowed us to dynamically confirm our static, confocal microscopy findings, while simultaneously enabling us to observe tumor cell-adherens junction interactions.

Fluorescent live-cell images of VE-cadherin-GFP-expressing HBMECs and CellTracker-stained MDA-MB-231 cells. (a) Example of MDA-MB-231 cell (white arrow) altering VE-cadherin-GFP (yellow arrow) between HBMECs as the tumor cell passes over the HBMEC junction. (b) Examples of MDA-MB-231 cells squeezing between HBMECs (pink and purple arrows) and possible material transfer from tumor cell to HBMEC, or potential transcellular incorporation (blue arrow). Intensity of each channel in each image was separately adjusted for better visualization, thus, relative intensities should not be compared. Scale bars from top row apply to all images in its series and are 50 μm. Starting time was selected for best visualization. All images were taken on HA/gelatin films with 0% Extralink. Green: VE-cadherin-GFP (HBMEC); red: CellTracker (MDA-MB-231).
MDA-MB-231 Tumor Cell Morphology and Migration Speed are Altered During Incorporation into HBMEC Monolayers
Breast tumor cell morphology and migration were significantly affected by the concentration of Extralink during migration on the bare HA/gelatin films (Figs. 1 and 2); therefore, we explored the dynamics of tumor cell incorporation into HBMECs and whether this process depended on crosslinking of the HA/gelatin films. In some cases, MDA-MB-231 cells either remained rounded on top of the HBMEC monolayer and did not incorporate, or they spread into the monolayer (Fig. 7a). However, many tumor cells also spread and migrated on top of the HBMEC monolayer without incorporating. Therefore, we studied three specific cases over time: (1) MDA-MB-231 cells that incorporated into the monolayer, separated into a pre-incorporation (i.e., exclusively rounded) and a post-incorporation (i.e., spread out) regime; (2) MDA-MB-231 cells that did not incorporate and remained rounded for the entire study; and (3) three representative HBMECs from each monolayer. First, we observed that the area (Fig. 10a) and speed (Fig. 10b) of HBMECs remained mostly constant over time at each Extralink concentration. There was no statistical difference between the absolute value of the average changes in HBMEC area or speed across all timepoints as a function of Extralink concentration (Fig. 10c). A similar trend was observed for non-incorporating MDA-MB-231 cells, where both their area (Fig. 10d) and speed (Fig. 10e) did not change significantly over time (Fig. 10f). In general, though, the greatest variability across timepoints in non-incorporating MDA-MB-231 cell area and speed occurred with the 1.2% Extralink concentration (Fig. 10f). However, the area of incorporating MDA-MB-231 cells increased during incorporation (Fig. 10g), while their speed decreased during incorporation (Fig. 10h). This increase in MDA-MB-231 cell size after incorporation is in line with a previous study that reported melanoma cells becoming more spread and elongated as they incorporated into endothelial monolayers.23 For tumor cells pre- and post-incorporation, there were no significant differences between Extralink concentrations in the absolute value of the change in area or speed of MDA-MB-231 cells across timepoints; however, it was evident that area was generally more consistent between the different Extralink concentrations prior to incorporation and speed was generally more consistent post-incorporation (Fig. 10i).

MDA-MB-231 cell area and speed profiles over time during incorporation into HBMEC monolayers. (a) HBMEC area and (b) HBMEC speed over time during incorporation experiments on HA/gelatin films with 0, 0.2, 0.8, 1.2, and 2% Extralink. 36 ≤ N ≤ 66 where N is number of cells. (c) Absolute value of the average change in HBMEC area and speed between each time point from panels (a) and (b), respectively. (d) Area and (e) speed over time of non-incorporating MDA-MB-231 cells on HBMEC monolayers on HA/gelatin films with 0, 0.2, 0.8, 1.2, and 2% Extralink. 14 ≤ N ≤ 89, where N is the number of cells. (f) Absolute value of the average change in MDA-MB-231 area and speed between each time point from panels (d) and (e), respectively. (g) Area and (h) speed over time for MDA-MB-231 cells pre-incorporation (while rounded) and post-incorporation (while spread out) into HBMEC monolayers. 30 ≤ N ≤ 78 where N is the number of cells. (i) Absolute values of the average change in MDA-MB-231 area and speed between each time point from panels (g) and (h), respectively. Average (j) area and (k) speed of HBMECs, non-incorporating MDA-MB-231 cells, and incorporating MDA-MB-231 cells (pre- and post-incorporation) on HA/gelatin films with 0, 0.2, 0.8, 1.2, and 2% Extralink. For panels (j) and (k), each dot represents one cell. All data is pooled from three independent trials. Statistical significance was determined using a non-parametric Kruskal–Wallis ANOVA test with a Dunn’s multiple comparison post-hoc test as follows: *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. For all figures, error signifies standard error of the mean.
To understand the statistical significance of this dynamic data, we calculated the area and speed of the MDA-MB-231 cells averaged across multiple time points pre- or post-incorporation. First, we found that there were no differences in HBMEC or MDA-MB-231 tumor cell (incorporating and non-incorporating) area (Fig. 10j) or speed (Fig. 10k) as a function of HA/gelatin film crosslinking under static conditions. However, the area of the MDA-MB-231 cells was significantly larger pre-incorporation compared to post-incorporation on HA/gelatin films for all concentrations of Extralink (Fig. 10j), as was also observed in the plots over time (Fig. 10g). Meanwhile, there was a significant decrease in speed between pre- and post-incorporation phases of the MDA-MB-231 cells on HA/gelatin films with 0, 0.8, and 2% Extralink (Fig. 10k). These results suggest that the process of incorporation into HBMECs changes the morphology and migratory behavior of the MDA-MB-231 cells and also abrogates the mechanosensitivity of the tumor cells to the crosslinking density of the HA/gelatin films.
We also note that for HA/gelatin films with 0.2% Extralink, the area of incorporating MDA-MB-231 cells (Fig. 10j) became similar to the area of MDA-MB-231 cells on bare films (Fig. 1b). However, due to the loss of a mechanosensation effect in the presence of HBMECs, MDA-MB-231 cells had much larger spreading areas and migration speeds during incorporation into HBMECs on the HA/gelatin films with 2.0% Extralink in comparison with MDA-MB-231 cells on the bare films.
Discussion
ECM crosslinking and stiffness are known regulators of many cellular functions in the context of physiologic and pathologic conditions. In this work, we explored the effects of HA crosslinking on metastatic breast tumor cell migration and incorporation into the human brain endothelium. Interestingly, our results showing reduced spreading area and more diffuse stress fibers for cells on more crosslinked, and thus stiffer, HA/gelatin films is contradictory to frequent reports in literature, including our own, where cells spread more on stiffer ECM-coated polyacrylamide gels.62,64,72 Similarly, previous studies have shown that when utilizing HA-methacrylate gels crosslinked with dithiothreitol (DTT) and functionalized with Arg-Gly-Asp (RGD) peptides for cell adhesion, increasing HA weight percent and crosslinking density, thus increasing stiffness, increased the spreading area and speed of glioblastoma cells.5 Also in opposition to our studies, Narkhede et al. found that increasing the concentration of DTT crosslinker in a methacrylated HA system with integrin binding protein coatings increased MDA-MB-231-BR cell area and speed.46 Notably, the work by Narkhede et al. was performed on HA gels formulated with different molecular weights, using a different mechanism of crosslinking, and with various coatings for cell adhesion, rather than gelatin incorporation as was used here. On the other hand, through chemical rather than mechanical signaling, soft HA gels can produce identical cellular responses to those on stiffer substrates.52 Hence, mechanosensitivity in this context likely involves a delicate balance between sensing of multiple parameters of the ECM, including stiffness, method and degree of crosslinking, method of adhesion molecule incorporation, and the specific composition of the matrix.
Our results suggest that HBMECs are less sensitive to this range of HA/gelatin crosslinking relative to MDA-MB-231 cells. This was not surprising, given that the measured Young’s modulus of the films was in the relatively small range of 0.85 to 3.8 kPa. We have previously shown that HBMEC morphology in a monolayer is sensitive to large differences in substrate stiffness (e.g., 194 kPa vs. approximately 50 GPa) but not to smaller ranges of stiffness (e.g., 1–194 kPa).25 Thus, from a mechanobiological perspective, the result that HBMEC morphology was not altered as a function of the Extralink concentrations is consistent with our expectations. The increase in overall ZO-1 presentation on the film made with 0.8% Extralink, including increased continuous junctions, suggests that the tight junctions of monolayers on this film composition may be more mature than those on the other films tested. Further investigation into how junction presentation of each individual junctional protein correlates with traditional barrier integrity assays (e.g., permeability, trans-endothelial electrical resistance) will help provide insight into the key players regulating barrier properties and provide measures for accepted characterization standards via JAnaP quantification in the future. However, we also note that an advantage of the JAnaP over traditional barrier integrity assays is the ability to quantitatively assess localized alterations in cell–cell junctions that may be present across heterogeneous monolayers. Furthermore, we expect that the addition of physiologic flow conditions into our assays will reveal new insights into the interplay between HA/gelatin crosslinking and shear stress effects on HBMEC morphology and junction presentation; this interplay may occur through a signaling crosstalk involving RhoA and EC nitric oxide synthase, as previously reported for bovine aortic ECs.35
Altogether, TCM minimally affected HBMEC size but altered ZO-1 and VE-cadherin junctional presentation to a more continuous and perpendicular morphology under these static conditions. Importantly, this increase in continuous junctions with TCM took the form of a jagged, zipper-like phenotype rather than a linear, mature continuous junction. This junctional data suggests that TCM treatment results in a shift of the monolayers to a less mature state, which appears to occur independently of matrix crosslinking. This shift in monolayer maturity was not surprising, since many reports have shown that metastatic breast tumor cell-secreted biochemical factors, such as VEGF, are capable of altering endothelial cells.8,37 Our results indicate that VEGF is in fact present in the TCM and that inhibiting its activity within TCM blocks the shift in junction presentation from a linear morphology to more perpendicular phenotype observed with TCM treatment. This is consistent with previous reports that VEGF induces a serrated junction presentation in HBMECs.37 Notably, most reports of junctional analysis rely on qualitative observation, fluorescence intensity image analysis, or quantification of the protein expression levels throughout the cells. An added benefit of using the JAnaP is the ability to simultaneously and semi-automatically quantify cell morphological parameters as well as the junctional proteins as they are presented at the cell–cell junction, providing a unique measurement capability to quantitatively compare between the TCM and TCM-free groups and assess heterogeneities in junctional morphology across the monolayer using statistical analysis. Together, these results suggest that the biochemical factors (e.g., VEGF) secreted by MDA-MB-231 cells alter HBMEC junction presentation in a manner unaffected by the extent of crosslinking of the underlying HA/gelatin film. It will be interesting to further examine this effect in the presence of physiologic flow conditions and in the presence of astrocytes and/or pericytes.
Our results suggest a lack of correlation between the number of tumor cells in the area of observation and propensity to incorporate. We note that if a tumor cell interacted directly with a gap between HBMECs in the monolayer, it was not included in the analysis. Therefore, once a monolayer of HBMECs was present in the system, breast tumor cells appeared to be less sensitive to HA/gelatin matrix crosslinking than when they were on bare films. These results suggest that HA/gelatin crosslinking does not significantly affect tumor cell incorporation into HMBEC monolayers, and that matrix mechanics might become more important post-extravasation, since tumor cells on the bare gels without HBMECs showed such dramatic differences in morphology and migration. Our observations for MDA-MB-231 and MDA-MB-231-BR cells incorporating into HBMEC monolayers on HA/gelatin films are also consistent with our previous results, where percent tumor cell incorporation into HUVECs was independent of polyacrylamide gel stiffness.63
Our observations suggest that breast tumor cells have multiple mechanisms by which they can interact with and incorporate into HBMEC monolayers. This is consistent with our prior studies in non-brain ECM, where tumor cells incorporated into and physically displaced endothelial cells in monolayers.27 Other studies have found that tumor cells can utilize cytoplasmic protrusions (i.e., invadopodia) to assist in extravasation across endothelial barriers and degrade the basement membrane,26,39,42,57 which has been previously thoroughly reviewed.20,61 It is possible that similar interactions are occurring here during paracellular incorporation as tumor cells squeeze in between brain endothelial cells. Furthermore, the dynamic nature by which we observed HBMEC adherens junctions disrupt due to tumor cell interaction and then reform is of particular interest, and suggest that HBMECs have a potential for self-repair. Indeed, our results are supported by prior reports where endothelial cells were able to rearrange their cytoskeleton to repair themselves post para- and trans-cellular invasion of tumor cells.7
Interestingly, other studies have reported enhanced transmigration of MDA-MB-231 cells in the presence of HBEMCs, suggesting that the ECs facilitate transmigration.38 This could potentially explain the increase in tumor cell area and migration post-incorporation, where HBMECs are present, compared to their respective values on bare HA/gelatin films. Notably, though, this response was influenced by the extent of matrix crosslinking, since only minimal changes were observed on the films with 0.2% Extralink. Alternatively, HBMECs may be remodeling the underlying matrix and/or depositing new ECM, hence masking the original mechanical properties of the HA/gelatin films to the incorporating tumor cells and altering their interactions with the matrix post-incorporation. Indeed, previous studies have demonstrated that ECs are capable of low levels of ECM deposition after just one day of culture in favorable conditions,28 and thus HBMEC-matrix remodeling is plausible during the 3-day culture period. It is also possible that cell adhesion molecules linking tumor cells and HBMECs could physically alter the migratory capacity of tumor cells within an HBMEC monolayer.
While our experiments have provided interesting insights into the biochemical and physical interactions between tumor cells, the HA matrix, and the brain endothelium, we acknowledge several important limitations of our system. First, our models lack other cell types present at the in vivo BBB, including astrocytes and pericytes. There is a growing body of literature15,30 to support the hypothesis that the presence of these cells, and/or their secreted biochemical factors, would likely influence tumor cell migration and incorporation into the brain endothelium, cell–cell junctions in the HBMECs, and possibly also the ECM. Indeed, our own previous work has shown that astrocyte conditioned media applied to MDA-MB-231 cells directly, or to their ECM only, can result in increased cell migration.60 Secondly, our models lack physiologic flow conditions. Shear stresses in the brain capillaries of rodent models have been measured to be in the range of 20–40 dyne/cm2,66 which can influence endothelial barrier function, cell migration, tumor cell adhesion, and tumor cell proliferation.3,9,16,36,48 The JAnaP can already calculate cell aspect ratios, and the next iteration of the program should include a feature that calculates alignment of endothelial cells to the direction of flow. Another limitation of our model is the lack of cylindrical geometry, which can influence EC elongation in the presence of shear stress.71 Incorporation of these additional cells and mechanical cues will continue to be important as the field develops benchmarks for BBB models.
Conclusions
We have shown that MDA-MB-231 and MDA-MB-231-BR (but not HBMEC) morphology and migration parameters are sensitive to the crosslinking density of HA/gelatin films, with a trend that is opposite of that typically observed for varying stiffness of ECM-coated polyacrylamide gels. Meanwhile, we used our novel JAnaP to quantify modest alterations in HBMEC tight and adherens junctions as a function of HA/gelatin crosslinking density. In addition, we found that tumor cell-secreted factors (e.g., VEGF) led to increased presentation of immature adherens junctions, but minimally affected tight junction presentation. HA/gelatin crosslinking, however, did not seem to affect MDA-MB-231 or MDA-MB_231-BR cell incorporation into HBMEC monolayers. We also showed that MDA-MB-231 tumor cells were able to utilize multiple modes of incorporation into HBMECs, in addition to the commonly reported paracellular pathway, and that MDA-MB-231 cells dynamically modified their morphology and migration behavior, as well as HBMEC junctions, during incorporation into the brain endothelium. Overall, our quantitative results suggest that a combination of biochemical and physical factors promote tumor cell migration through the brain endothelium, and suggest that ECM mechanics may become most significant once incorporation or transmigration is complete. Future work will continue to build upon these results by incorporating more cellular components and mechanical cues into the experimental in vitro models.
Abbreviations
- AFM:
-
Atomic force microscopy
- ANOVA:
-
Analysis of variance
- AR:
-
Aspect ratio
- BBB:
-
Blood-brain barrier
- BSA:
-
Bovine serum albumin
- DMEM:
-
Dulbecco’s modified eagle’s medium
- DMSO:
-
Dimethyl sulfoxide
- ECGS:
-
Endothelial cell growth supplement
- ECM:
-
Extracellular matrix
- FBS:
-
Fetal bovine serum
- GFP:
-
Green fluorescent protein
- HA:
-
Hyaluronic acid
- HBMEC:
-
Human brain microvascular endothelial cell
- JanaP:
-
Junction Analyzer Program
- LOX:
-
Lysyl oxidase
- PBS:
-
Phosphate buffered saline
- RPMI:
-
Roswell Park Memorial Institute
- STR:
-
Short tandem repeat
- TCM:
-
Tumor conditioned media
- TJ:
-
Tight junctions
- VE-cadherin:
-
Vascular endothelial cadherin
- VEGF:
-
Vascular Endothelial Growth Factor
- ZO-1:
-
Zonula occludens-1
References
Abbott, N. J., and A. Friedman. Overview and introduction: the blood-brain barrier in health and disease. Epilepsia 53(Suppl 6):1–6, 2012.
Achrol, A. S., R. C. Rennert, C. Anders, R. Soffietti, M. S. Ahluwalia, L. Nayak, S. Peters, N. D. Arvold, G. R. Harsh, P. S. Steeg, and S. D. Chang. Brain metastases. Nat. Rev. Dis. Primers 5:5, 2019.
Ajami, N. E., S. Gupta, M. R. Maurya, P. Nguyen, J. Y.-S. Li, J. Y.-J. Shyy, Z. Chen, S. Chien, and S. Subramaniam. Systems biology analysis of longitudinal functional response of endothelial cells to shear stress. Proc. Natl. Acad. Sci. 114:10990–10995, 2017.
Akiri, G., E. Sabo, H. Dafni, Z. Vadasz, Y. Kartvelishvily, N. Gan, O. Kessler, T. Cohen, M. Resnick, M. Neeman, and G. Neufeld. Lysyl oxidase-related protein-1 promotes tumor fibrosis and tumor progression in vivo. Cancer Res. 63:1657–1666, 2003.
Ananthanarayanan, B., Y. Kim, and S. Kumar. Elucidating the mechanobiology of malignant brain tumors using a brain matrix-mimetic hyaluronic acid hydrogel platform. Biomaterials 32:7913–7923, 2011.
Arshad, F., L. Wang, C. Sy, S. Avraham, and H. K. Avraham. Blood-brain barrier integrity and breast cancer metastasis to the brain. Patholog. Res. Int. 1–12:2010, 2011.
Arvanitis, C., S. Khuon, R. Spann, K. M. Ridge, T.-L. Chew, and L. Kreplak. Structure and biomechanics of the endothelial transcellular circumferential invasion array in tumor invasion. PLoS ONE 9:e89758, 2014.
Avraham, H. K., S. Jiang, Y. Fu, H. Nakshatri, H. Ovadia, and S. Avraham. Angiopoietin-2 mediates blood-brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J. Pathol. 232:369–381, 2014.
Baeyens, N., C. Bandyopadhyay, B. G. Coon, S. Yun, and M. A. Schwartz. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Invest. 126:821–828, 2016.
Barnes, J. M., L. Przybyla, and V. M. Weaver. Tissue mechanics regulate brain development, homeostasis and disease. J. Cell Sci. 130:71–82, 2017.
Bellail, A. C., S. B. Hunter, D. J. Brat, and E. G. Van Meir. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 36:1046–1069, 2004.
Cai, J., W. G. Jiang, and R. E. Mansel. Phosphorylation and disorganization of vascular-endothelial cadherin in interaction between breast cancer and vascular endothelial cells. Int. J. Mol. Med. 4:191–195, 1999.
Chen, W., A. D. Hoffmann, H. Liu, and X. Liu. Organotropism: new insights into molecular mechanisms of breast cancer metastasis. Precis. Oncol. 2:4, 2018.
Cox, T. R., and J. T. Erler. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis. Model. Mech. 4:165–178, 2011.
Destefano, J. G., J. J. Jamieson, R. M. Linville, and P. C. Searson. Benchmarking in vitro tissue-engineered blood-brain barrier models. Fluids Barriers CNS 15:32, 2018.
DeStefano, J. G., Z. S. Xu, A. J. Williams, N. Yimam, and P. C. Searson. Effect of shear stress on iPSC-derived human brain microvascular endothelial cells (dhBMECs). Fluids Barriers CNS 14:20, 2017.
Discher, D. E., P. Janmey, and Y.-L. Wang. Tissue cells feel and respond to the stiffness of their substrate. Science 310:1139–1143, 2005.
Dorland, Y. L., and S. Huveneers. Cell-cell junctional mechanotransduction in endothelial remodeling. Cell. Mol. Life Sci. 74:279–292, 2017.
Dun, M. D., R. J. Chalkley, S. Faulkner, S. Keene, K. A. Avery-Kiejda, R. J. Scott, L. G. Falkenby, M. J. Cairns, M. R. Larsen, R. A. Bradshaw, and H. Hondermarck. Proteotranscriptomic profiling of 231-BR breast cancer cells: identification of potential biomarkers and therapeutic targets for brain metastasis. Mol. Cell. Proteomics 14:2316–2330, 2015.
Eddy, R. J., M. D. Weidmann, V. P. Sharma, and J. S. Condeelis. Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends Cell Biol. 27:595–607, 2017.
Eichler, A. F., E. Chung, D. P. Kodack, J. S. Loeffler, D. Fukumura, and R. K. Jain. The biology of brain metastases-translation to new therapies. Nat. Rev. Clin. Oncol. 8:344–356, 2011.
Fan, J., and B. M. Fu. Quantification of malignant breast cancer cell MDA-MB-231 transmigration across brain and lung microvascular endothelium. Ann. Biomed. Eng. 44:2189–2201, 2016.
Fazakas, C., I. Wilhelm, P. Nagyoszi, A. E. Farkas, J. Haskó, J. Molnar, H. Bauer, H.-C. Bauer, F. Ayaydin, N. T. K. Dung, L. Siklós, and I. A. Krizbai. Transmigration of melanoma cells through the blood-brain barrier: role of endothelial tight junctions and melanoma-released serine proteases. PLoS ONE 6:e20758, 2011.
Grammas, P., J. Martinez, and B. Miller. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev. Mol. Med. 13:e19, 2011.
Gray, K. M., D. B. Katz, E. G. Brown, and K. M. Stroka. Quantitative phenotyping of cell-cell junctions to evaluate ZO-1 presentation in brain endothelial cells. Ann. Biomed. Eng. 2019. https://doi.org/10.1007/s10439-019-02266-5.
Hagedorn, E. J., J. W. Ziel, M. A. Morrissey, L. M. Linden, Z. Wang, Q. Chi, S. A. Johnson, and D. R. Sherwood. The netrin receptor DCC focuses invadopodia-driven basement membrane transmigration in vivo. J. Cell Biol. 201:903–913, 2013.
Hamilla, S. M., K. M. Stroka, and H. Aranda-Espinoza. VE-Cadherin-independent cancer cell incorporation into the vascular endothelium precedes transmigration. PLoS ONE 9:e109748, 2014.
Hielscher, A., K. Ellis, C. Qiu, J. Porterfield, and S. Gerecht. Fibronectin deposition participates in extracellular matrix assembly and vascular morphogenesis. PLoS ONE 11:e0147600, 2016.
Hoshino, A., et al. Tumour exosome integrins determine organotropic metastasis. Nature 527:329–335, 2015.
Jamieson, J. J., P. C. Searson, and S. Gerecht. Engineering the human blood-brain barrier in vitro. J. Biol. Eng. 11:37, 2017.
Kass, L., J. T. Erler, M. Dembo, and V. M. Weaver. Mammary epithelial cell: Influence of extracellular matrix composition and organization during development and tumorigenesis. Int. J. Biochem. Cell Biol. 3(39):1987–1994, 2007.
Katt, M. E., R. M. Linville, L. N. Mayo, Z. S. Xu, and P. C. Searson. Functional brain-specific microvessels from iPSC-derived human brain microvascular endothelial cells: the role of matrix composition on monolayer formation. Fluids Barriers CNS 15:7, 2018.
Kienast, Y., L. Von Baumgarten, M. Fuhrmann, W. E. F. Klinkert, R. Goldbrunner, J. Herms, and F. Winkler. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 16:116–122, 2010.
Kim, Y., and S. Kumar. CD44-mediated adhesion to hyaluronic acid contributes to mechanosensing and invasive motility. Mol. Cancer Res. 12:1416–1429, 2014.
Kohn, J. C. C., D. W. W. Zhou, F. Bordeleau, A. L. L. Zhou, B. N. N. Mason, M. J. J. Mitchell, M. R. R. King, and C. A. A. Reinhart-King. Cooperative effects of matrix stiffness and fluid shear stress on endothelial cell behavior. Biophys. J. 108:471–478, 2015.
Lee, H. J., M. F. Diaz, K. M. Price, J. A. Ozuna, S. Zhang, E. M. Sevick-Muraca, J. P. Hagan, and P. L. Wenzel. Fluid shear stress activates YAP1 to promote cancer cell motility. Nat. Commun. 8:14122, 2017.
Lee, T.-H., H. Karsenty Avraham, S. Jiang, and S. Avraham. Vascular endothelial growth factor modulates the transendothelial migration of MDA-MB-231 breast cancer cells through regulation of brain microvascular endothelial cell permeability. J. Biol. Chem. 278:5277–5284, 2003.
Lee, K. Y., Y.-J. Kim, H. Yoo, S. H. Lee, J. B. Park, and H. J. Kim. Human brain endothelial cell-derived COX-2 facilitates extravasation of breast cancer cells across the blood-brain barrier. Anticancer Res. 31:4307–4313, 2011.
Leong, H. S., A. E. Robertson, K. Stoletov, S. J. Leith, C. A. Chin, A. E. Chien, M. N. Hague, A. Ablack, K. Carmine-Simmen, V. A. Mcpherson, C. O. Postenka, E. A. Turley, S. A. Courtneidge, A. F. Chambers, and J. D. Lewis. Article invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 8:1558–1570, 2014.
Levental, K. R., H. Yu, L. Kass, J. N. Lakins, M. Egeblad, J. T. Erler, S. F. T. Fong, K. Csiszar, A. Giaccia, W. Weninger, M. Yamauchi, D. L. Gasser, and V. M. Weaver. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139:891–906, 2009.
Li, B., W.-D. Zhao, Z.-M. Tan, W.-G. Fang, L. Zhu, and Y.-H. Chen. Involvement of Rho/ROCK signalling in small cell lung cancer migration through human brain microvascular endothelial cells. FEBS Lett. 580:4252–4260, 2006.
Mader, C. C., M. Oser, M. A. O. Magalhaes, J. J. Bravo-Cordero, J. Condeelis, A. J. Koleske, and H. Gil-Henn. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. J. Cancer Res. 71:OF1–OF12, 2011.
Martin, T. A., and W. G. Jiang. Loss of tight junction barrier function and its role in cancer metastasis. Biochim. Biophys. Acta Biomembr. 1788:872–891, 2009.
McFarlane, S., J. A. Coulter, P. Tibbits, A. O’Grady, C. McFarlane, N. Montgomery, A. Hill, H. O. McCarthy, L. S. Young, E. W. Kay, C. M. Isacke, and D. J. J. Waugh. CD44 increases the efficiency of distant metastasis of breast cancer. Oncotarget 6:11465–11476, 2015.
Mouw, J. K., G. Ou, and V. M. Weaver. Extracellular matrix assembly: a multiscale deconstruction. Nat. Publ. Gr. 15:771, 2014.
Narkhede, A. A., J. H. Crenshaw, R. M. Manning, and S. S. Rao. The influence of matrix stiffness on the behavior of brain metastatic breast cancer cells in a biomimetic hyaluronic acid hydrogel platform. J. Biomed. Mater. Res. A 106:1832–1841, 2018.
Nayak, L., E. Q. Lee, and P. Y. Wen. Epidemiology of brain metastases. Curr. Oncol. Rep. 14:48–54, 2012.
Northcott, J. M., I. S. Dean, J. K. Mouw, and V. M. Weaver. Feeling stress: the mechanics of cancer progression and aggression. Front. Cell Dev. Biol. 6:17, 2018.
Novak, U., and A. H. Kaye. Extracellular matrix and the brain: components and function. J. Clin. Neurosci. 7:280–290, 2000.
Onken, M. D., J. Li, and J. A. Cooper. Uveal melanoma cells utilize a novel Route for transendothelial migration. PLoS ONE 9:e115472, 2014.
Onken, M. D., O. L. Mooren, S. Mukherjee, S. T. Shahan, J. Li, and J. A. Cooper. Endothelial monolayers and transendothelial migration depend on mechanical properties of the substrate. Cytoskeleton 71:695–706, 2014.
Pogoda, K., R. Bucki, F. J. Byfield, K. Cruz, T. Lee, C. Marcinkiewicz, and P. A. Janmey. Soft substrates containing hyaluronan mimic the effects of increased stiffness on morphology, motility, and proliferation of glioma cells. Biomacromolecules 18:3040–3051, 2017.
Prestwich, G. D., and C. O. N. Spectus. Evaluating drug efficacy and toxicology in three dimensions: using synthetic extracellular matrices in drug discovery. Acc. Chem. Res. 41:139–148, 2008.
Reymond, N., P. Riou, and A. J. Ridley. Rho GTPases and cancer cell transendothelial migration. Methods Mol. Biol. 827:123–142, 2012.
Roberts, H. C., T. P. L. Roberts, R. C. Brasch, and W. P. Dillon. Quantitative measurement of microvascular permeability in human brain tumors achieved using dynamic contrast-enhanced mr imaging: correlation with histologic grade. AJNR Am. J. Neuroradiol. 21:891–899, 2000.
Rodriguez, P. L., S. Jiang, Y. Fu, S. Avraham, and H. K. Avraham. The proinflammatory peptide substance P promotes blood-brain barrier breaching by breast cancer cells through changes in microvascular endothelial cell tight junctions. Int. J. Cancer 134:1034–1044, 2014.
Roh-Johnson, M., J. J. Bravo-Cordero, A. Patsialou, V. P. Sharma, P. Guo, H. Liu, L. Hodgson, and J. Condeelis. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 33:4203–4212, 2014.
Sarrió, D., S. M. Rodriguez-Pinilla, D. Hardisson, A. Cano, G. Moreno-Bueno, and J. Palacios. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 68:989–997, 2008.
Shaw, S. K., P. S. Bamba, B. N. Perkins, and F. W. Luscinskas. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J. Immunol. 167:2323–2330, 2001.
Shumakovich, M. A., C. P. Mencio, J. S. Siglin, R. A. Moriarty, H. M. Geller, and K. M. Stroka. Astrocytes from the brain microenvironment alter migration and morphology of metastatic breast cancer cells. FASEB J. 31:5049–5067, 2017.
Sibony-Benyamini, H., and H. Gil-Henn. Invadopodia: the leading force. Eur. J. Cell Biol. 91:896–901, 2012.
Stroka, K. M., and H. Aranda-Espinoza. Neutrophils display biphasic relationship between migration and substrate stiffness. Cell Motil. Cytoskelet. 66:328–341, 2009.
Stroka, K. M., H. N. Hayenga, and H. Aranda-Espinoza. Human neutrophil cytoskeletal dynamics and contractility actively contribute to trans-endothelial migration. PLoS ONE 8:61377, 2013.
Stroka, K. M., B. Sheng Wong, M. Shriver, J. M. Phillip, D. Wirtz, A. Kontrogianni-Konstantopoulos, and K. Konstantopoulos. Loss of giant obscurins alters breast epithelial cell mechanosensing of matrix stiffness. Oncotarget 5:54004–54020, 2016.
Tornavaca, O., M. Chia, N. Dufton, L. O. Almagro, D. E. Conway, A. M. Randi, M. A. Schwartz, K. Matter, and M. S. Balda. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 208:821–838, 2015.
Turitto, V. T. Blood viscosity, mass transport, and thrombogenesis. Prog. Hemost. Thromb. 6:139–177, 1982.
Vallenius, T. Actin stress fibre subtypes in mesenchymal-migrating cells. Open Biol. 3:130001, 2013.
Vanderhooft, J. L., M. Alcoutlabi, J. J. Magda, and G. D. Prestwich. Rheological properties of cross-linked hyaluronan-gelatin hydrogels for tissue engineering. Macromol. Biosci. 2009. https://doi.org/10.1002/mabi.200800141.
Wrobel, J. K., and M. Toborek. Blood–brain barrier remodeling during brain metastasis formation. Mol. Med. 22:32–40, 2016.
Yankaskas, C. L., K. N. Thompson, C. D. Paul, M. I. Vitolo, P. Mistriotis, A. Mahendra, V. K. Bajpai, D. J. Shea, K. M. Manto, A. C. Chai, N. Varadarajan, A. Kontrogianni-Konstantopoulos, S. S. Martin, and K. Konstantopoulos. A microfluidic assay for the quantification of the metastatic propensity of breast cancer specimens. Nat. Biomed. Eng. 2019. https://doi.org/10.1038/s41551-019-0400-9.
Ye, M., H. M. Sanchez, M. Hultz, Z. Yang, M. Bogorad, A. D. Wong, and P. C. Searson. Brain microvascular endothelial cells resist elongation due to curvature and shear stress. Sci. Rep. 4:4681, 2014.
Yeung, T., P. C. Georges, L. A. Flanagan, B. Marg, M. Ortiz, M. Funaki, N. Zahir, W. Ming, V. Weaver, and P. A. Janmey. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskelet. 60:24–34, 2005.
Yoneda, T., P. J. Williams, T. Hiraga, M. Niewolna, and R. Nishimura. A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J. Bone Miner. Res. 16:1486–1495, 2001.
Zhang, P., C. Fu, H. Bai, E. Song, and Y. Song. CD44 variant, but not standard CD44 isoforms, mediate disassembly of endothelial VE-cadherin junction on metastatic melanoma cells. FEBS Lett. 588:4573–4582, 2014.
Zheng Shu, X., Y. Liu, F. S. Palumbo, Y. Luo, and G. D. Prestwich. In situ crosslinkable hyaluronan hydrogels for tissue engineering. Biomaterials 25:1339–1348, 2004.
Acknowledgments
We thank Dr. Toshiyuki Yoneda for generously providing MDA-MB-231-BR cells. The University of Maryland Computer, Mathematical, and Natural Sciences imaging incubator is acknowledged for providing training and equipment for confocal imaging. Kyle Thomas at Yellow Basket, LLC (kyle@yellowbasket.io) is acknowledged for the JAnaP software development support. We also acknowledge Mary Doolin for help with editing custom Matlab code. We thank Dr. William Luscinskas from the Harvard Medical School for generously providing us with the VE-cadherin-GFP adenovirus.
Funding
Funding was provided by Burroughs Wellcome Fund (Career Award at the Scientific Interface). Additional funding was provided by the Ann G. Wylie Dissertation Fellowship from the University of Maryland Graduate School (to MAP), the Fischell Fellowship in Biomedical Engineering (to KMG), the Dr. Mabel S. Spencer Award for Excellence in Graduate Achievement (to KMG), the Clark Doctoral Fellowship (to AJD), the Fischell Department of Bioengineering, and the University of Maryland.
Author Contributions
KMS, MAP, and KMG designed the research. MAP and GMD performed experiments for Fig. 1. GMD analyzed all data for Fig. 1. AJLD performed all experiments and data analysis for Fig. 2, with guidance from MAP. MAP performed confocal microscopy for Fig. 3. KMG performed experiments and analysis for Figs. 4, 5, 6, S2, S3, and S4, with help in analysis from JWJ. KMG prepared Fig. S1. MAP performed experiments and analysis for Figs. 7a–7e. AJLD performed experiments for Figs. 7f–7h, with guidance from MAP, and MAP analyzed data for Figs. 7f–7h. MAP performed confocal microscopy for Fig. 8. MAP and AJLD performed experiments for Fig. 9. MAP performed experiments and all analysis for Fig. 10. MAP performed statistical analysis for Figs. 1, 2, 7, and 10. MAP formatted Figs. 1, 2, 3, 7, 8, 9, and 10. KMG performed statistical analysis and/or formatting for Figs. 4, 5, 6, S2, S3, and S4. MAP, KMG, and KMS wrote the manuscript. All authors edited the manuscript, and all authors reviewed and approved final version of the manuscript.
Conflict of interest
MAP, KMG, GMD, AJLD, JWJ, and KMS declare that they have no conflict of interest.
Human Studies
No human studies were carried out by the authors for this article.
Animal Studies
No animal studies were carried out by the authors for this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Michael King oversaw the review of this article.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kimberly Stroka, in January 2015, joined the Fischell Department of Bioengineering as an Assistant Professor at the University of Maryland, College Park. She received her B.S. in Physics in 2006 from Denison University and her PhD in Bioengineering from the University of Maryland, College Park. She completed her postdoctoral training at Johns Hopkins University in the Department of Chemical and Biomolecular Engineering and Institute for NanoBioTechnology. Dr. Stroka has received the National Science Foundation Graduate Research Fellowship, NIH NRSA F31 predoctoral fellowship, NIH T32 and F32 postdoctoral fellowships, and Burroughs Wellcome Career Award at the Scientific Interface. She also received the Rita Schaffer Young Investigator Award from the Biomedical Engineering Society (2014), Research and Scholarship Award from the UMD Graduate School (2017), “Outstanding Young Scientist Award” from the Maryland Academy of Sciences (2017), and Fischell Department of Bioengineering Faculty Teaching Award (2018). Dr. Stroka’s lab engineers cells and their microenvironment in order to create model systems that allow them to systematically understand fundamental aspects of cellular and tissue mechanobiology, with applications in vascular biomechanics, tumor cell metastasis, and stem cell engineering.

This article is part of the CMBE 2019 Young Innovators special issue.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pranda, M.A., Gray, K.M., DeCastro, A.J.L. et al. Tumor Cell Mechanosensing During Incorporation into the Brain Microvascular Endothelium. Cel. Mol. Bioeng. 12, 455–480 (2019). https://doi.org/10.1007/s12195-019-00591-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12195-019-00591-2