
Abstract
microRNAs (miRNAs) are a class of small non-coding RNAs that play key roles in post transcriptional gene regulation that influence various fundamental cellular processes, including the cellular responses during environmental stresses. However, perusal of literatures revealed few reports on the differential expression of miRNA during thermal stress in Indian native (Bos indicus) cattle breeds. The present investigation aimed to identify differentially expressed miRNAs during thermal stress in Sahiwal (Bos indicus) dairy cattle breed of India, adapted with tropical climate over a long period of time. Stress responses of the animals were characterized by determining various physiological as well as biochemical parameters and differential expression profile of major heat shock protein genes. Ion Torrent deep sequencing and CLC-genomic analysis identified a set of differentially expressed miRNAs during summer and winter seasons. Most of the identified differentially expressed miRNAs were found to target heat shock responsive genes especially members of heat shock protein (HSP) family. Real-time quantification-based analysis of selected miRNAs revealed that bta-mir-1248, bta-mir-2332, bta-mir-2478, and bta-mir-1839 were significantly (p < 0.01) over expressed while bta-mir-16a, bta-let-7b, bta-mir-142, and bta-mir-425 were significantly (p < 0.01) under expressed during summer in comparison to winter. The present study enlists differentially expressed miRNAs at different environmental temperatures in Sahiwal (Bos indicus) that may be importance for further understanding the role of miRNAs on thermo-regulatory mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Surrounding environment plays a significant role in livestock productivity. Among the different environmental factors, thermal stress seems to be one of the most fascinating factor affecting livestock production as well as health. In tropical, subtropical, and arid regions, high ambient temperature is one of the major factors affecting high producing animal production (Krishnan et al. 2017). Heat tolerance can be defined as “ability of the body to endure the impact of a hot environment without suffering ill-effects” (Bianca 1961). In comparison to temperate environments, indigenous animals reared in tropical or arid regions are more adapted to hot climate (Najjar et al. 2010).
micro RNA (miRNA) are a class of small non-coding RNAs with the size of 22 base pairs, play a significant roles in biotic as well as abiotic stresses by regulating their target genes (Leung and Sharp 2010; Song et al. 2017; Sengar et al. 2018). In total, more than 60% of mammalian protein coding genes are under the selective pressure to pair with miRNAs (Friedman et al. 2009). Thermo stability and dynamics of miRNA are also significant in its proper functioning as well as interaction with other molecular factors to maintain different physiological conditions (Paul et al. 2013; Bharatiy et al. 2016). Multiple lines of genetic studies were undertaken to rule out the function of miRNA during stress (Xu et al. 2003; van Rooij et al. 2007; Li et al. 2009; Flynt et al. 2007; Leung and Sharp 2010). Knockout of miR-7 and miR-14 from flies not able to develop eyes and have increased sensitivity towards salinity, respectively, during abnormal environmental situations (Li et al. 2009; Xu et al. 2003). miR-208-deficient mice are unable to cope with cardiac overload (van Rooij et al. 2007). Inactivation of miR-8 in zebrafish can make them unresponsive to osmotic pressures (Flynt et al. 2007). Stress conditions can change the bio-genesis of miRNAs, expression profile of mRNA targets as well as activities of miRNA-protein complexes (Leung and Sharp 2010). miRNAs perform multiple tasks during stress responses (Leung and Sharp 2010). p53 can regulate the expression of specific miRNAs either at transcription or processing levels. p53 induces the transcription of primary transcripts that encompass miR-34a, miR-34b, and miR-34c from distinct genomic loci at the time of DNA destruction and consequently, a number of genes targeted by these miRNAs are suppressed resulting in growth arrest as well as apoptosis (Hermeking 2007). Apart from the cellular concentration of miRNAs, target gene repression is also dependent on the mRNA targets concentration relative to the miRNA (Doench et al. 2003), amount of other mRNAs present in the transcriptome that are targeted by the same miRNA (Franco-Zorrilla et al. 2007). In addition to the relative concentration of miRNA as well as mRNA targets, the regulatory properties of miRNA depend on the properties of other RNA-binding proteins that can interact with nearby miRNA-protein complexes on the same mRNA target (Leung and Sharp 2010). In some situations, miRNAs act as regulators of stress responses and is particularly important during acute stress responses such as inflammation. In response to inflammation, miR-9, miR-155, and miR-146 transcripts are upregulated along with other inflammatory-responsive genes through a signaling cascade in macrophages (O’Connell et al. 2010).
At present, little information is available on the differential expression pattern of various miRNAs during thermal stress in different cattle breeds (Zheng et al. 2014; Muroya et al. 2015; Sengar et al. 2018). In the present study, we catalogued a number of differentially expressed miRNAs during summer in Sahiwal (Bos indicus) cattle, breed, that will be interesting for screening of miRNA-based biomarkers related to thermal stress in cattle.
Materials and methods
Animals and sample collection
Experimental protocols including animals were permitted by the Institutional Animal Ethics Committee (IAEC), ICAR-CIRC, Meerut, Uttar Pradesh, India. Ten animals of Sahiwal (Bos indicus) cattle breed maintained at the same managemental regimen were randomly selected. Blood samples were collected from the same experimental animals during winter (January–February; temperature ranges 15–18 °C) and summer (May–June; temperature ranges 42–45 °C) seasons. Temperature humidity index (THI) was used as an indicator of heat stress, regardless of whether the animals appeared heat stressed. Prior to collection of the samples, animals were exposed to the natural environmental temperatures for 1 h, and when the environmental temperature was within the range of 42–45 °C (summer) or 15–18 °C (winter), collection was done at 2:00 PM (Sengar et al. 2018). Stress responses of animals were determined by recording physiological parameters, and doing biochemical assays and expression profiling of two major stressor genes (HSP70 and HSP90). Physiological parameters viz. rectal temperature (RT), respiratory rate, and pulse rate were recorded on days of sampling at both seasons (Kurz et al. 2013).
Determination of biochemical parameters
Peripheral blood mononuclear cells (PBMC) were isolated from the collected blood samples followed by separation of plasma as per the protocols described earlier (Deb et al. 2013; Deb et al. 2014; Sengar et al. 2018). Isolated PBMC samples were preserved in liquid nitrogen (− 196 °C) until total RNA isolation. Antioxidant levels (nmol/min/ml) in plasma samples were detected using catalase (CAT) and glutathione peroxidase (GPx) assay kits (Cayman chemical, USA). Thiobarbituric acid reactive substances (TBARS) assay (Cayman chemical, USA) was conducted for colorimetric measurement of malondialdehyde (MDA) (μM), a product of lipid peroxidation.
Detection of HSP70 and HSP90 transcripts
Total RNA was isolated from the PBMC samples using cold trizol method (Sigma Aldrich, USA) and quantified through spectrophotometrically. Integrity was determined by agarose gel visualization of 18 s and 28 s ribosomal bands. Isolated total RNA was subjected for cDNA synthesis using ProtoScript first strand cDNA synthesis kit (New England Biolabs, Beverly, MA, USA). Diluted cDNA (1:10) samples were used for Real-time qPCR (Step One, Applied Biosystems, Foster City, CA, USA) based quantification. Readers may retrieve the primer sequences for HSP70, HSP90, and beta-actin gene (endogenous control) from our previous studies (Deb et al. 2014; Bhanuprakash et al. 2016; Sengar et al. 2018). Real-time PCR reaction was carried out in a final reaction volume of 10 μl via SYBR Green® PCR Master Mix kit (Applied Biosystems, Foster City, CA, USA), according to manufacturer’s instructions. ΔΔCt method was applied for quantification of the samples (Livak and Schmittgen 2001). All the reactions were performed in triplicate and the detected expression values were normalized against the endogenous control.
Small RNA enrichments and RNA integrity number determination
Pooled PBMC samples of each season were subjected for total RNA isolation using PAX gene blood miRNA kit (Qiagen, USA) according to the manufacturer’s instructions. Quality and quantity of purified enriched small RNAs were determined through NanoDrop™ Spectrophotometer and Qubit fluorometer (Life technologies, USA). RNA integrity was determined using Agilent™ Small RNA Kit (Agilent, USA) in RNA 6000 pico chip as per manufacturer’s instructions through Agilent™ 2100 Bioanalyzer™ instrument. RNA integrity numbers (RINs) were detected using 2100 expert software and the highest-quality library mapping statistics were obtained from input RNA with higher RIN values.
Construction of small RNA libraries and deep sequencing
Small RNA libraries were constructed using total RNA-Seq kit v2 for small RNA libraries kit (Thermo Fisher scientific, USA) according to the manufacturer’s instruction. One microliter of the purified cDNA library was run on an Agilent™ 2100 Bioanalyzer™ instrument with the Agilent™ DNA 1000 kit. Smear analysis was performed using the 2100 expert software to determine the molar concentration of cDNA libraries having size range of 50–300 bp. Each of the barcoded cDNA libraries was diluted to the same molar concentration (nM) and the pool of barcoded libraries was prepared by mixing all of them. The Ion XPress Template kit V2.0 (Life Technologies, USA) was used to perform emulsion PCR as per the user guidelines (Part No. 4469004 Rev. B 07/2011). Deep sequencing of the cDNA libraries was performed using Ion Torrent Personal Genome Machine (Life Technologies, Pleasanton, CA, USA).
Deep sequencing data analysis
Obtained raw sequences were passed through CLC–Genomics-work bench 8.0.2 software (Qiagen, Denmark) to eliminate the adapter and low-quality sequences. Extracted small RNA sequences with the range of 15–26 nucleotides were processed for RFam and Repbase filter, further the remaining sequences were compared with miRBase 21 by BLASTn search to identify the conserved Bos taurus miRNAs (http://www.mirbase.org/). For identification of the potential precursor miRNA sequences, three maximum mismatches were allowed between the identified miRNAs and known animal miRNAs, followed by processing of the identified all mature miRNA sequences for BLAST against the downloaded bovine draft genome sequences (http://www.ncbi.nlm.nih.gov/genome/82) and the hairpin RNA structures for their flanking sequences were determined by RNA fold software (http://mfold.rit.albany.edu/?q=mfold/RNA-Folding-Form).
Real-time PCR-based validation of differential expression of miRNAs
Based on the deep sequencing results, differentially expressed eight miRNAs (bta-mir-1248, bta-mir-2332, bta-mir-2478, bta-mir-1839, bta-mir-16a, bta-let-7b, bta-mir-142, and bta-mir-425) were selected for validation through real-time quantitative PCR (RT-qPCR). Total RNA was isolated using PAX gene blood miRNA kit (Qiagen, USA) followed by reverse transcription using a protoscript first strand cDNA synthesis kit (New England Biolabs, Beverly, MA, USA) as per the manufacturer’s recommendation. Diluted cDNA samples were used to carry out the RT-qPCR. The miRNA oligonucleotides synthesized for the quantification had the similar sequences as the Bos taurus miRNA with suitable adjustments at their 5′ ends. The PCR reaction mixtures were performed in a final reaction volume of 10 μl using the SYBR Green® PCR Master Mix kit (Applied Biosystems, Foster City, CA, USA), according to manufacturer’s recommendation. Normalization of the relative expression values of each miRNAs was performed against beta-actin, an endogenous control gene, permitting the comparison of samples independently of the amount of total input template (Sengar et al. 2018). Triplicate replicas were performed for all the reactions and quantification of the samples was done by the ΔΔCt method (Livak and Schmittgen 2001).
Prediction of target genes
Target Scan software (Lewis et al. 2003) was used for prediction of the target mRNA related to heat stress, mostly the heat shock proteins. Further, combining the target gene function database with miRNA and mRNA sequence using miRNA-GO-network analysis, it was identified that multiple genes were simultaneously regulated by the same miRNA.
miRNA network analysis and functional annotation by gene ontology
Network-based visual analysis of miRNA, their targets, and functions were performed by using miRNet (http://www.mirnet.ca/faces/home.xhtml) as per the web-based approach developed by Fan et al. (2016). miRNet provides two well-established node centrality measures to estimate node importance—degree centrality and betweenness centrality. In a graph network, the degree of a node (miRNA) is the number of connections it has to other nodes. Nodes with higher node degree act as hubs in a network. The betweenness centrality measures the number of shortest paths going through the node. Network visualization and functional enrichment analysis is a powerful combination to deliver important biological insight. miRNet supports functional annotations based on Gene Ontology (GO) or KEGG databases. For miRNA input, an enrichment algorithm has been implemented in miRNet called hypergeometric tests as described by Bealzard et al. (2015).
Statistical analysis
Data are presented as mean ± SEM and analyzed by using SPSS statistical program (SPSS 10.0 for Windows; SPSS, Inc., Chicago, IL, USA). Significant differences were determined by one-way ANOVA using the SPSS program.
Results
Characterization of stress response—physiological parameters and biochemical assay
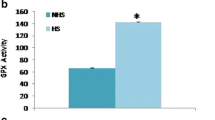
Stress response was characterized by recording different physiological parameters viz. rectal temperature, respiratory rate, and pulse rate at two different seasons were recorded. Table 1 represents that all the physiological parameters were comparatively higher during summer, indicating that the animals were under reasonable thermal stress. Further, we also conducted certain biochemical assays for characterizing stress response in summer. Plasma catalase (CAT) activity (nmol/min/ml) during summer (22.365 ± 0.76) was significantly (p < 0.01) higher than winter season (2.519 ± 0.76) (Fig. 1a). Similarly, a significant (p < 0.01) increased level of GPx (nmol/min/ml) was determined in the plasma samples collected during summer (145.201 ± 1.26) in comparison to the winter (61.263 ± 1.26) (Fig. 1b). Calculated plasma concentration of MDA (μM) was 0.011 ± 0.003 and 0.058 ± 0.003 during winter and summer seasons, respectively. A significant (p < 0.01) difference in the MDA concentration was observed between the seasons (Fig. 1c).

Antioxidants and lipid peroxidation by-products in the plasma samples of Sahiwal cattle collected during winter and summer. a Catalase (CAT) activity (nmol/min/ml); b glutathione peroxidase (GPx) level (nmol/min/ml) and c malondialdehyde (MDA) concentration (μM); **p < 0.01
Expression profile of stressor genes
Differential relative expression (mean ± SE) level of both HSP70 and HSP90 transcripts was significantly (p < 0.01) higher during summer season in comparison to winter (Fig. 2).

Relative mRNA expression (mean ± standard error) of HSP90 and HSP70 in the Sahiwal cattle PBMC during winter and summer seasons. **p < 0.01
Deep sequencing
Existence of small RNAs during post enrichment was observed (Fig. 3a). Twenty to 25 nucleotides ranges of small RNAs including microRNAs were detected on the gel showing a great quantity of small RNAs and slighter intensity of ribosomal RNA bands. Detected RIN were in the ranges of 5.8–6.5 (Fig. 3b). There were 46,938 and 39,925 raw reads in the small RNA libraries of summer and winter season groups, respectively, with a length of 14–30 nucleotides after filtering of the adapter as well as other low-quality sequences. Majority of small RNAs were 20~25 nucleotides in length among both the libraries. Bos taurus miRBase v20 database was used for mapping of small RNA libraries. Annotated miRNAs during summer and winter were 150 and 172, respectively (Table 2). miRNAs percentage in the total small RNA libraries of summer and winter were 50 and 53%, respectively. Figure 4 represents the increase in expression level of miRNAs with varying color intensity in terms of reads per kilo millions (RPKM) values, and based on the change of color intensity, the differentially expressed miRNAs were selected for validation. miRNAs fold changes in the two small RNA libraries were analyzed as per the methodology described earlier by Zheng et al. (2014) (Table 3). Table 4 represents the target genes of the selected miRNAs and most were heat shock protein coding genes.

Assessment of small RNA enrichments (a) Agilent 2100 bioanalyzer electrophoresis of RNA samples at pre and post enrichments (enriched small RNA are shown in green round). L, RNA ladder; 1, winter post enrichment; 2, summer post enrichment; 3, winter pre enrichment and 4, summer post enrichment. b RNA integrity numbers (RIN) obtained through bioanalyzer reading after small RNA enrichments

Heat map for differentially expressed miRNAs during winter (left) vs summer (right). The rows are centered using vector scaling and they are clustered using Euclidean distance and complete linkage. Intensity of the different colors indicates variation in the level of expression for a particular miRNA during winter vs summer seasons. Same or different color in both the column indicates null or maximum variation of expression levels, respectively. The columns are clustered using correlation distance and average linkage. There are total 57 rows, 2 columns
Real-time PCR-based validation of the selected miRNA expression
Real-time qPCR-based validation revealed a significant (p < 0.01) difference in the expression levels of the respective miRNAs during summer and winter seasons that was in agreement with the normalized deep sequencing results (Figs. 5 and 6). Among the selected miRNAs, bta-mir-2478 was found to be highly expressed (p < 0.01) during summer compared to the winter season.

RT-PCR-based profiling of top four highly expressed miRNAs during summer in Sahiwal cattle. a Expression of miRNAs by ion torrent deep sequencing (normalized reads). b Based relative expression of miRNAs. The results revealed that expression of these selected RT-qPCR miRNAs by RT-qPCR was in agreement with the normalized sequencing data.*p < 0.01

RT-PCR-based profiling of top four lowly expressed miRNAs during summer in Sahiwal cattle. a Expression of miRNAs by ion torrent deep sequencing (normalized reads). b Based relative expression of miRNAs. The results revealed that expression of these selected RT-qPCR miRNAs by RT-qPCR were in agreement with the normalized sequencing data.*p < 0.01
miRNA network analysis
As per the centrality analysis methods described by Koschützki and Schreiber (2008), we have mapped the miRNA network of bta-mir-7b, bta-mir-16a, bta-mir-1248, bta-mir-2478, bta-mir-2332, and bta-mir-1839. Among these miRNAs, bta-mir-16a has the highest degree centrality because it interacts with highest number of other miRNAs. Betweenness centrality of bra-mir-16a was also found to be highest among the other miRNAs. bta-mir-7b miRNA shows the second highest degree centrality and betweenness centrality. bta-mir-1248 and bta-mir-2478 have the degree of centrality with other miRNA of 6 and 3, respectively. bta-mir-1839 has the lowest degree centrality and betweenness of 4 and 6, respectively, among all (Fig. 7).

miRNA network analysis by miRNet with the values of their respective degree centrality and betweenness centrality. Here, the degree of centrality talks about the node having the highest connections in the network that act as hubs. Thus finding such nodes suggests essential genes
Gene ontology-based network analysis
Functional annotation of the targeted miRNAs of Sahiwal cattle was generated using the KEGG ontology database. Network analysis showed that the target miRNA HSD17B12 and TAF6 are the two most clustered when interacting with bta-mir-1248 from Sahiwal cattle. HSD17B12 and TAF6 miRNAs are responsible for maintaining metabolic pathways and Herpes simplex infection, respectively. Other functions such as fatty acid elongation, O-glycan biosynthesis, unsaturated fatty acid biosynthesis, and fatty acid metabolism are regulated by the miRNA HSD17B12 and MFNG that also interact with bta-mir-1248. TAF6 acts as basal transcription factor shown to have interaction with bta-mir-1248. MFNG miRNA was also responsible for maintaining the Notch signaling pathway. HSD17B12 miRNA has the second most clustering probability when it acts as a steroid hormone synthesis regulator. Tested miRNA bta-mir-1839 has interactions with another miRNA called POLL that controls non-homologous end-joining and base excision repair. These two functional annotations recorded least according to the probability scale (Table 5). Figure 8 represents the Venn diagram on ontology analysis through the KEGG database of functional genes that are targeted by the tested miRNA. The diagram depicts the probability (0–1) of gene showing the particular function where it is a measure of total involvement of the miRNA for a particular function; 1 is highest and the involvement will be decreased as the value comes down from 1 towards 0.

The Venn diagram representing ontology analysis through the KEGG database of functional genes that are targeted by tested miRNA. The diagram shows probability of gene (range: 0–1) showing the particular function, where it is measure of total involvement of the miRNA for a particular function; 1 is highest and the involvement will be decreased as the value comes down from 1 towards 0. Each section in diagram represent the particular miRNA’s enriched function (respective targeted miRNAs are in brackets)
Discussion
High environmental temperatures directly or indirectly affects the productivity of dairy cattle. Similarly, heavy beef cattle are unable to withstand thermal stress compared to lighter cattle because high fat deposition averts heavy cattle from regulating their body temperature efficiently. Upon exposure to adverse conditions such as heat shock, hypoxia, and metabolic/oxidative stresses, translation of cellular mRNAs is tightly regulated to allow rapid adaptation to environmental changes. miRNAs are known post transcriptional regulators. Multiple genetic studies have investigated the post transcriptional regulatory mechanisms of miRNA during stresses (Islam et al. 2014; Place and Noonan 2014). However, there are very few reports regarding the role of miRNAs during environmental stresses in bovine species (Zheng et al. 2014; Muroya et al. 2015; Sengar et al. 2018). Recently, we identified 65 differentially expressed miRNAs during elevated environmental temperatures in crossbred (Bos taurus × Bos indicus) cattle (Sengar et al. 2018). In the present study, we aimed to catalogue differentially expressed miRNAs during summer stress among Sahiwal cattle (Bos indicus), an Indian native milch breed. Due to their long-term association with tropical conditions, Indian native cattle breeds are known to have higher heat stress resilience compared to the taurine or cross bred strains.
The stress response was characterized by physiological as well as biochemical parameters. Physiological parameters including respiratory rate, heart beats, and body/skin temperature provide a direct measure of climatic stress (Roman-Ponce et al. 1977; Davies and Maconochie 2009). More recently, the animal’s ability to tolerate the climatic stress under temperate conditions has been determined physiologically by assessing the changes in body temperature, respiratory, and pulse rate (Charoensook et al. 2012; Deb et al. 2013). The present study showed that summer environmental stress can upshift the rectal temperature, respiratory, and pulse rate in comparison to winter seasons in the Sahiwal cattle breed.
Heat stress is one of the major factors causing oxidative stress during summer in animals. Heat stress results when the core body temperature rises above its normal range, resulting from a total heat load exceeding the capacity for heat dissipation (Ganaie et al. 2013). Rising global concern of thermal discomfort in farm animals is debatable not only for countries of tropical zones, but also for nations of temperate zones where ambient temperatures are increasing due to climate change (Nardone et al. 2010). Complex biological antioxidant systems sustain homeostasis in changing environments by providing defense against oxidative stress to the aerobic organisms (Bagnyukova et al. 2007). Our earlier studies reported that heat stress can significantly enhance the antioxidant enzymes in crossbred cattle (Sengar et al. 2018). Profiling of two antioxidant enzymes during thermal stress in Sahiwal cattle revealed significantly (p < 0.01) increased activities of catalase (8.8-fold) and glutathione peroxidase (2.2-fold) during summer in comparison to the winter seasons. Similarly, catalase and glutathione peroxidase concentrations were significantly higher during summer in comparison to the winter seasons among growing calves, heifers, and lactating Murrah buffaloes (Lallawmkimi 2009). During thermal stress due to augmented SOD activity, hydrogen peroxide production was found to increase (Bernabucci et al. 2002) which in turn boosted the plasma catalase as well as glutathione peroxidase concentrations (Hazra et al. 2009). Heat stress can affect lipid metabolism and antioxidant activity during lactation as well as reproductive performance in heifers/primiparous cows (Turk et al. 2015). Similar to the recent report by Sengar et al. (2018), we measured the lipid peroxidation by product, TBARS/MDA concentration during summer and winter seasons in Sahiwal cattle. MDA concentration was significantly (p < 0.01) higher during summer than winter seasons. The present study is in agreement with earlier findings that environmental heat stress can raise both of TBARS and MDA levels in broilers (Mujahid et al. 2007), dairy cows (Chandra and Agarwal 2009; Aengwanich et al. 2011; Sengar et al. 2018), and buffalo (Belhadj Slimen et al. 2016).
Cellular thermo-tolerance is mediated by a family of heat shock proteins (Hsp), among them, Hsp70 and Hsp90 are the most abundant as well as temperature-sensitive chaperones (Craven et al. 1996; Beckham et al. 2004; Wandinger et al. 2008). Considering the major indicators of stress response, we characterized the HSP70 and HSP90 expression in the PBMC of Sahiwal cattle during summer and winter seasons. Results are in agreement with our earlier reports (Deb et al. 2014; Bhanuprakash et al. 2016; Sengar et al. 2018).
miRNAs are differentially expressed during heat stress in taurine or crossbred cattle (Zheng 2014; Sengar et al. 2018). Circulating miRNAs have been identified in the grazing cattle (Muroya et al. 2015). The level of miR-150, miR-19b, miR-148a, miR-221, miR-223, miR-320a, miR-361, and miR-486 was temporarily lower in the cattle after 1 and/or 2 months of grazing compared to housed cattle, while the level of miR-451 was upregulated in grazing cattle (Muroya et al. 2015). Expression profile of miR-19a, miR-19b, miR-27b, miR-30a-5p, miR-181a, miR-181b, miR-345-3p, and miR-1246 was comparatively higher in the serum of heat-stressed than the non-heat-stressed Holstein cows (Zheng et al. 2014). Sixty-five identified differentially expressed miRNAs in the PBMC of crossbred cattle, among them bta-miR-103-2, bta-miR-2898, bta-miR-2478, and bta-miR-181b-2, were significantly overexpressed during thermal stress (Sengar et al. 2018). The present study identified a number of differentially expressed miRNAs in Sahiwal (Bos indicus) breed, during seasonal variations. Quantitative validation of the selected miRNAs revealed that bta-mir-1248, bta-mir-2332, bta-mir-2478, and bta-mir-1839 were significantly (p < 0.01) over expressed during summer in comparison to winter. However, bta-mir-16a, bta-let-7b, bta-mir-142, and bta-mir-425 were significantly (p < 0.01) under expressed during summer compared to the winter. Interestingly, bta-mir-2478 was also over expressed in crossbreed cattle during summer season (Sanger et al. 2018). Mir-2478 plays an important role in regulation of bovine energy metabolism and/or adipogenesis (Romao et al. 2014). Upregulation of mir-2478 in bovine muscle is associated with the expression of Stearoyl-CoA desaturase-1 (SCD1), a lipogenic gene (Muroya et al. 2016).
According to miRNA network analysis, we have found that the bta-mir-16a is important in sense of constructing maximum interaction with other miRNAs. bta-mir-7b is also shown to possess near similar significance according to number of interacting partners. Similar approaches using gene ontology through KEGG database has studied multifunctional transcription factor (TF) gene EWS/EWSR1 involved in various cellular processes such as transcription regulation, noncoding RNA regulation, splicing regulation, genotoxic stress response, and cancer generation (Lee et al. 2016). In our study, gene ontology prediction using the KEGG database of the selected miRNAs implied the importance of bta-mir-1248 and bta-mir-1839. These two miRNAs were found to be mainly responsible for regulating different metabolic pathways of heat stress response and these two miRNAs have the most cross-talks with other miRNA. Interactome studies from gene ontology determination showed that bta-mir-1839 miRNA have crucial impact on the regulation of POLL miRNA that induce proper non-homologous end-joining and base excision repair. In order to preserve the integrity of DNA, cells utilized DNA repair mechanisms especially base excision repair pathway (Sancar et al. 2004; Friedberg et al. 2006). Oxidative stress alters base excision repair pathway and increases apoptotic response (Unnikrishnan et al. 2009). Being an inducer of DNA repair mechanisms, POLL miRNA may play an important role to control cellular integrity during stressful conditions.
From these findings, we observe that almost all the miRNAs are related to different metabolic pathways and target various genes that are involved in a broad spectrum of metabolic as well as physiological processes. Variation in the expression level of miRNAs at different seasons may differentially regulate the metabolic process and thus may be associated with thermal stress.
Our present findings provide a list of differentially expressed miRNAs at different environmental temperatures in Sahiwal cattle, an Indian native (Bos indicus) breed that may add some references towards developing miRNA-based biomarkers for selecting thermo resilience cattle to a dairy herd.
References
Aengwanich W, Kongbuntad W, Boonsorn T (2011) Effects of shade on physiological changes, oxidative stress, and total antioxidant power in Thai Brahman cattle. Int J Biometeorol 55:741–748
Bagnyukova TV, Lushchak OV, Storey KB, Lushchak VI (2007) Oxidative stress and antioxidant defense responses by goldfish tissues to acute change of temperature from 3 to 23°C. J Therm Biol 32:227–234
Beckham JT, Mackanos MA, Crooke C, Takahashi T, O’Connell-Rodwell C, Contag CH, Jansen ED (2004) Assessment of cellular response to thermal laser injury through bioluminescence imaging of heat shock protein 70. Photochem Photobiol 79:76–85
Belhadj Slimen I, Najar T, Ghram A, Abdrrabba M (2016) Heat stress effects on livestock: molecular, cellular and metabolic aspects, a review. J Anim Physiol Anim Nutr 100(3):401–412
Bernabucci U, Ronchi B, Lacetera N, Nardone A (2002) Markers of oxidative status in plasma and erythrocytes of transition dairy cows during hot season. J Dairy Sci 85(9):2173–2179
Bhanuprakash V, Singh U, Sengar G, Sajjanar B, Bhusan B, Raja TV, Alex R, Kumar S, Singh R, Kumar A, Alyethodi RR, Kumar S, Deb R (2016) Differential effect of thermal stress on Hsp70 expression, nitric oxide production and cell proliferation among native and crossbred dairy cattle. J Therm Biol 59:18–25
Bharatiy S, Hazra M, Paul M, Mohapatra S, Samantaray D, Dubey RC, Sanyal S, Datta S, Hazra S (2016) In silico designing of an industrially sustainable carbonic anhydrase using molecular dynamics simulation. ACS Omega 1:1081–1103
Bianca W (1961) Heat tolerance in cattle-its concept, measurement and dependence on modifying factors. Int J Biometeorol 5(1):5–30
Chandra G, Aggarwal A (2009) Effect of DL-α-tocopherol acetate on calving induced oxidative stress in periparturient crossbred cows during summer and winter seasons. Indian J Anim Nutr 26:204–210
Charoensook R, Gatphayak K, Sharifi AR, Chaisongkram C, Brenig B, Knorr C (2012) Polymorphisms in the bovine HSP90AB1 gene are associated with heat tolerance in Thai indigenous cattle. Trop Anim Health Prod 44(4):921–928
Craven RA, Egerton M, Stirling CJ (1996) A novel Hsp70 of the yeast ER lumen is required for the efficient translocation of a number of protein precursors. EMBO J 15:2640–2650
Davies P, Maconochie I (2009) The relationship between body temperature, heart rate and respiratory rate in children. Emerg Med J 26(9):641–643
Deb R, Sajjanar B, Singh U, Kumar S, Brahmane MP, Singh R, Sengar G, Sharma A (2013) Promoter variants at AP2 box region of Hsp70. 1 affects thermal stress response and milk production traits in Frieswal cross bred cattle. Gene 532(2):230–235
Deb R, Sajjanar B, Singh U, Kumar S, Singh R, Sengar G, Sharma A (2014) Effect of heat stress on the expression profile of Hsp90 among Sahiwal (Bos indicus) and Frieswal (Bos indicus× Bos taurus) breed of cattle: a comparative study. Gene 536(2):435–440
Doench JG, Petersen CP, Sharp PA (2003) siRNAs can function as miRNAs. Genes Dev 17(4):438–442
Fan Y, Siklenka K, Arora SK, Ribeiro P, Kimmins S, Xia J (2016) miRNet—dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acids Res 44:135–141
Flynt AS, Li N, Thatcher EJ, Solnica-Krezel L, Patton JG (2007) Zebrafish miR-214 modulates hedgehog signaling to specify muscle cell fate. Nat Genet 39:259–263
Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva WA, García DJA, Paz-Ares J (2007) Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet 39(8):1033–1037
Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T (2006) DNA repair and mutagenesis. ASM Press, Washington D.C, pp 169–226
Friedman RC, Farh KKH, Burge CB, Bartel DP (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19(1):92–105
Ganaie AH, Shanker G, Bumla NA, Ghasura RS, Mir NA, Wani SA, Dudhatra GB (2013) Biochemical and physiological changes during thermal stress in bovines. J Vet Sci Technol 4(126):126–132
Hazra S, Sabini E, Ort S, Konrad M, Lavie A (2009) Extending thymidine kinase activity to the catalytic repertoire of human deoxycytidine kinase. Biochemistry 48:1256–1263
Hermeking H (2007) p53 enters the microRNA world. Cancer Cell 12(5):414–418
Islam M, Datta J, Lang JC, Teknos TN (2014) Down regulation of RhoC by microRNA-138 results in de-activation of FAK, Src and Erk 1/2 signaling pathway in head and neck squamous cell carcinoma. Oral Oncol 50(5):448–456
Koschützki D, Schreiber F (2008) Centrality analysis methods for biological networks and their application to gene regulatory networks. Gene Regul Syst Biol 2:193–201
Krishnan G, Bagath M, Pragna P, Vidya MK, Aleena J, Archana PR, Sejian V, Bhatta R (2017) Mitigation of the heat stress impact in livestock reproduction. In: Theriogenology. InTech. https://doi.org/10.5772/INTECHOPEN.69091
Kurz S, Wolff K, Hazra S, Bethel C, Hujer A, Smith K, Xu Y, Tremblay L, Blanchard JS, Nguyen L, Bonomo R (2013) Can inhibitor resistant substitutions in the Mycobacterium tuberculosis β-lactamase BlaC lead to clavulanate resistance? A biochemical rationale for the use of β-lactam β-lactamase inhibitor combinations. Antimicrob Agents Chemother 57:6085–6096
Lallawmkimi CM (2009) Impact of thermal stress and vitamin-E supplementation on heat shock protein 72 and antioxidant enzymes in Murrah buffaloes. Deemed University, Karnal (Haryana), India, Ph.D.Thesis submitted to National Dairy Research Institute
Lee C-J, Ahn H, Lee SB, Shin J-Y, Park W-Y, Jong-Il K, Lee J, Ryu H, Kim S (2016) Integrated analysis of omics data using microRNA-target mRNA network and PPI network reveals regulation of Gnai1 function in the spinal cord of Ews/Ewsr1 KO mice. BMC Med Genet 9(Suppl 1):33
Leung AKL, Sharp PA (2010) MicroRNA functions in stress responses. Mol Cell 40(2):205–215
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115:787–798. https://doi.org/10.1016/S0092-8674(03)01018-3
Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW (2009) A microRNA imparts robustness against environmental fluctuation during development. Cell 137(2):273–282
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25(4):402–408
Mujahid A, Pumford NR, Bottje W, Nakagawa K, Miyazawa T, Akiba Y, Toyomizu M (2007) Mitochondrial oxidative damage in chicken skeletal muscle induced by acute heat stress. Poult Sci 44:439–445
Muroya S, Hagi T, Kimura A, Aso H, Matsuzaki M, Nomura M (2016) Lactogenic hormones alter cellular and extracellular microRNA expression in bovine mammary epithelial cell culture. J Anim Sci Biotechnol 7(1):8
Muroya S, Ogasawara H, Hojito M (2015) Grazing affects exosomal circulating microRNAs in cattle. PLoS One 10(8):e0136475
Najjar RG, Pyke CR, Adams MB, Breitburg D, Hershner C, Kemp M, Howarth R, Mulholland MR, Paolisso M, Secor D, Sellner K (2010) Potential climate-change impacts on the Chesapeake Bay. Estuar Coast Shelf Sci 86(1):1–20
Nardone A, Ronchi B, Lacetera N, Ranieri MS, Bernabucci U (2010) Effects of climate changes on animal production and sustainability of livestock systems. Livest Sci 130(1):57–69
O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D (2010) MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 33(4):607–619
Paul M, Hazra M, Barman A, Hazra S (2013) Comparative molecular dynamics simulation studies for determining factors contributing to the thermostability of chemotaxis protein Che Y. J Biomol Struct Dyn 32:928–949
Place RF, Noonan EJ (2014) Non-coding RNAs turn up the heat: an emerging layer of novel regulators in the mammalian heat shock response. Cell Stress and Chaperones 19(2):159–172
Romao JM, Jin W, He M, McAllister T (2014) MicroRNAs in bovine adipogenesis: genomic context, expression and function. BMC Genomics 15(1):137
Roman-Ponce H, Thatcher WW, Buffington DE, Wilcox CJ, Van Horn HH (1977) Physiological and production responses of dairy cattle to a shade structure in a subtropical environment. J Dairy Sci 60(3):424–430
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85
Sengar GS, Deb R, Singh U, Raja TV, Kant R, Sajjanar B, Alex R, Alyethodi RR, Kumar A, Kumar S, Singh R (2018) Differential expression of microRNAs associated with thermal stress in Frieswal (Bos taurus x Bos indicus) crossbred dairy cattle. Cell Stress and Chaperones 23:155–170. https://doi.org/10.1007/s12192-017-0833-6
Song G, Zhang R, Zhang S, Li Y, Gao J, Han X, Chen M, Wang J, Li W, Li G (2017) Response of microRNAs to cold treatment in the young spikes of common wheat. BMC Genomics 18:212. https://doi.org/10.1186/s12864-017-3556-2
Turk R, Podpecan O, Mrkun J, Flegar-Mestri Z, Perkov S, Zrimsek P (2015) The effect of seasonal thermal stress on lipid mobilisation, antioxidant status and reproductive performance in dairy cows. Reprod Domest Anim 50:595–603. https://doi.org/10.1111/rda.12534
Unnikrishnan A, Raffoul JJ, Patel HV, Prychitko TM, Anyangwe N, Meira LB, Friedberg EC, Cabelof DC, Heydari AR (2009) Oxidative stress alters base excision repair pathway and increases apoptotic response in apurinic/apyrimidinic endonuclease 1/redox factor-1 haploinsufficient mice. Free Radic Biol Med 46(11):1488–1499
van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN (2007) Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316(5824):575–579
Wandinger SK, Richter K, Buchner J (2008) The Hsp90 chaperone machinery. J Biol Chem 283(27):18473–18477
Xu P, Vernooy SY, Guo M, Hay BA (2003) The Drosophila microRNA Mir-14 suppresses cell death and is required for normal fat metabolism. Curr Biol 13(9):90–795
Zheng Y, Chen K, Zheng X, Li H, Wang G (2014) Identification and bioinformatics analysis of microRNAs associated with stress and immune response in serum of heat-stressed and normal Holstein cows. Cell Stress Chaperones 19(6):973–981. https://doi.org/10.1007/s12192-014-0521-8
Acknowledgements
The authors are thankful to the Director, ICAR-CIRC for providing necessary facilities to carry out the present research. We are also thankful to Military Farm, Meerut, India for providing experimental animals; Ome Research Laboratory, Anand Agricultural University, Gujrat, India for NGS analysis.
Funding
The authors acknowledge the Science and Engineering Research Board, Government of India for providing financial support under the project YSS/2014/000279 to RD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declared that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Sengar, G.S., Deb, R., Singh, U. et al. Identification of differentially expressed microRNAs in Sahiwal (Bos indicus) breed of cattle during thermal stress. Cell Stress and Chaperones 23, 1019–1032 (2018). https://doi.org/10.1007/s12192-018-0911-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-018-0911-4