
Abstract
Atrial fibrillation (AF) is the most common tachyarrhythmia which is associated with increased morbidity and mortality. AF usually progresses from a self-terminating paroxysmal to persistent disease. It has been recognized that AF progression is driven by structural remodeling of cardiomyocytes, which results in electrical and contractile dysfunction of the atria. We recently showed that structural remodeling is rooted in derailment of proteostasis, i.e., homeostasis of protein production, function, and degradation. Since heat shock proteins (HSPs) play an important role in maintaining a healthy proteostasis, the role of HSPs was investigated in AF. It was found that especially small heat shock protein (HSPB) levels get exhausted in atrial tissue of patients with persistent AF and that genetic or pharmacological induction of HSPB protects against cardiomyocyte remodeling in experimental models for AF. In this review, we provide an overview of HSPBs as a potential therapeutic target for normalizing proteostasis and suppressing the substrates for AF progression in experimental and clinical AF and discuss HSP activators as a promising therapy to prevent AF onset and progression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
AF progression by structural remodeling
Atrial fibrillation (AF) is an age-related tachyarrhythmia in both left and right atria, which can be caused by underlying (heart) conditions, such as valvular heart disease, congestive heart disease, ischemic cardiomyopathy, obesity, diabetes mellitus, and hypertension (Dobrev et al. 2012; Hoogstra-Berends et al. 2012). The goal of AF therapy is, ideally, to abolish AF episodes and to restore normal sinus rhythm. Unfortunately, treatment of AF remains difficult, which is caused by the persistent and progressive nature of this arrhythmia. There are strong indications that remodeling of the structure of atrial cardiomyocytes underlies electrophysiological and contractile dysfunction and AF perpetuation (de Groot et al. 2010). Structural remodeling includes degradation of sarcomeres (the smallest contractile units of the cardiomyocytes), namely myolysis, by proteases such as calpain (Brundel et al. 2002; Ke et al. 2008) and disruption of the microtubule network (Zhang et al. 2014), which result in impaired electrical coupling and functional recovery to sinus rhythm after pharmacological and electrical cardioversion (Ausma et al. 1997; Kirubakaran et al. 2015; Todd et al. 2004). Importantly, structural changes are already presented when a patient enters the clinic, for the first time, with an episode of AF. Since the current available therapy is directed at alleviation of electrophysiological changes (rhythm control), it has limited effect on patient’s outcome. Therapeutic approaches that counteract the pathways conveying AF-induced structural remodeling may offer superior therapeutic perspectives. Recent research findings indicate that derailment of proteostasis, i.e., the homeostasis of protein production, function, and degradation, constitutes an important factor for the induction and progression of AF. In addition, it was observed that especially small heat shock proteins (HSPBs) convey protective effects against derailment of proteostasis and thereby attenuate structural remodeling, AF onset, and progression.
Proteostasis and role for HSPs
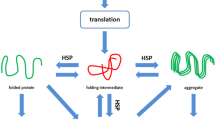
It has been recognized that a proper function of cells and organisms depends critically on the maintenance of a healthy proteostasis (Balch et al. 2008; Kampinga and Bergink 2016). Proteostasis is particularly important in long-lived post-mitotic cardiomyocytes, since they display limited regenerative capacity. Proteostasis involves controlling the concentration, conformation, binding interaction, kinetics, and location of individual proteins. Derailment of cellular proteostasis results in many systemic diseases, including cardiovascular disorders (Balch et al. 2008). Cells respond to a loss of proteostatic control by inducing a heat shock response (HSR), upon which HSPs are expressed. Interestingly, in the heart, numerous HSPs are already expressed at high levels, especially the family of small HSP members: HSPB1, HSPB5, HSPB6, HSPB7, and HSPB8. These HSPBs are considered to constitute the cell’s first line of defense against proteostasis derailment by stabilizing the sarcomere (Brundel et al. 2006a; Ke et al. 2008). In general, HSPs act as molecular chaperones to facilitate protein folding, localization, degradation, and function, thereby maintaining proteostasis and preventing various forms of cardiomyocyte damage (Westerheide and Morimoto 2005). Indeed, HSPs were found to play a protective role in various cardiovascular diseases, including AF. Two studies reported induced expression of mitochondrial HSPs, HSPD1, HSPE1, and mortalin (HSPA9B) in atrial tissue of patients with AF. These HSPs may play a protective role by maintaining mitochondrial integrity and capacity for ATP generation (Kirmanoglou et al. 2004; Schafler et al. 2002). Unfortunately, no mechanistic studies have been performed to conclusively address their function. Other studies revealed induced HSPA1A expression in atrial tissue of patients undergoing cardiac surgery. Higher HSPA1A expression correlated with lower incidence of post-surgery AF, suggesting a cardioprotective role for HSPA1A (Mandal et al. 2005; St Rammos et al. 2002). A key role for HSPB members in the protection against AF onset and progression was identified by several studies (Brundel et al. 2006a, b; Ke et al. 2011; Zhang et al. 2011). Interestingly, it was found that overexpression of HSPB1 protects against contractile dysfunction by conservation of the cardiomyocyte structure in the tachypaced HL-1 cardiomyocyte model for AF and in clinical AF (Brundel et al. 2006a), suggesting HSPB1 to represent a druggable target in AF.
Key role for HSPB members in the prevention of cardiac diseases
HSPB members
The family of HSPBs consists of at least ten members, and they are expressed in various human tissues (Vos et al. 2009) (Table 1). HSPB members are defined by a conserved C-terminal domain of approximately 90 amino acids (the α-crystallin domain) flanked by a variable length N-terminal arm and a more conserved C-terminal extension (Bakthisaran et al. 2015). Some HSPB members, including HSPB1, HSPB5, and HSPB8, are thought to assemble into homo- and/or heterogeneous oligomeric complexes, which dissociate into smaller multimers upon stress. Another important characteristic is that various HSPB members can be phosphorylated, which changes their activity and oligomeric state (Vos et al. 2008).
HSPBs have at least three, not mutually exclusive, biochemical functions within the proteostasis regulation. Firstly, both in vitro and in vivo findings suggested that some HSPB members act as ATP-independent chaperones by preventing irreversible protein aggregation (Carra et al. 2009; Chowdary et al. 2004; Sanbe et al. 2007). Oligomeric dynamics seem to be crucial for such HSPB activities (van Montfort et al. 2001). HSPB clients may also be processed (renaturation or degradation) through cooperation with ATP-dependent chaperones (Mogk et al. 2003; Veinger et al. 1998). As such, stress-induced protein damage, which may also occur in AF, can be prevented. Secondly, several HSPB members are associated with cytoskeletal proteins in a phosphorylation-dependent manner (Golenhofen et al. 2004; Landry and Huot 1995; Lavoie et al. 1995). This results in stabilization of cytoskeletal structures and increased resistance to stress situations, including AF. Finally, HSPB members are found to inhibit the activation of proteases and as such may prevent the activation of calpain, which was found to become activated in clinical AF (Brundel et al. 2002; Zhang et al. 2011).
Relevant HSPB family members for heart function: functional similarities and divergence
Various HSPB members are expressed at high levels in the heart (Golenhofen et al. 2004; Verschuure et al. 2003) (Table 1). One of these members is HSPB1. HSPB1 can exist as high or low molecular weight structures. Under normal, non-stressed conditions, a high molecular weight structure is the most predominant form. During proteotoxic stress, its level decreases whereas the level of phosphorylated low molecular weight structures increases (Vos et al. 2008). In addition to the role of HSPB1 in assisting in refolding and/or targeting denatured proteins, another well-studied role of HSPB1 is its ability to interact with several cytoskeletal proteins, including actin, intermediate filaments, and microtubules (Landry and Huot 1995; Vicart et al. 1998). In heart tissue, HSPB1 is found associated with sarcomeres and thereby was found to be cardioprotective (Brundel et al. 2006a).
HSPB2 associates specifically with dystrophy myotonic protein kinase (DMPK) and therefore is called a DMPK-binding protein, indicating its importance in muscle maintenance (Kadono et al. 2006; Suzuki et al. 1998). It is highly expressed in heart and skeletal muscle and was found to have protective effects against cardiac diseases, such as cardiac hypertrophy and ischemia heart diseases (Ishiwata et al. 2012; Nakagawa et al. 2001; Sugiyama et al. 2000). Also, HSPB2 was found to be associated with the outer membrane of mitochondria, thereby regulating the mitochondria permeability transition and calcium uptake in mitochondria. Overexpression of HSPB2 was found to conserve ATP synthesis in mice with ischemic/reperfusion injury (Nakagawa et al. 2001). Mice with specific knockout of HSPB2 show, upon ischemic stress, reduced mitochondria respiration rates and ATP production as well as suppression in expression of several metabolic and mitochondrial regulators (Ishiwata et al. 2012). These findings imply that HSPB2 is cardioprotective via maintenance of mitochondrial function and metabolic activity during cardiac stress. This role has been confirmed in a study utilizing a double knockout of HSPB2 and HSPB5. Here, inhibition of mitochondrial calcium signaling and, consequently, a reduction in ATP synthesis were observed during ischemia/reperfusion (Kadono et al. 2006). Findings from the study of Golenhofen et al. imply that the increased calcium in the cytosol, due to knockout of HSPB2, may modify the calcium sensitivity of myofibrils, contributing to malfunction of cardiac contractility (Golenhofen et al. 2006). Interestingly, mice overexpressing cardiac HSPB2 revealed lower levels of cardiac biomarker troponin I in the blood after ischemia/reperfusion injury, indicating that troponin I levels in heart tissue are conserved, thereby preserving contractile function of the heart (Grose et al. 2015).
HSPB3 and HSPB4 are not expressed in the heart (Vos et al. 2009), whereas HSPB5 co-localizes on the I-band and M-line region of sarcomeres in cardiomyocytes (van de Klundert et al. 1998). HSPB5 is known to bind and stabilize intermediate filaments, actin microfilaments, and sarcomeric proteins, including actin, desmin, and titin (Bullard et al. 2004; Ghosh et al. 2007; Perng et al. 1999). Like HSPB1, HSPB5 also plays an important role in stabilization of the cytoskeleton as it is expressed together with HSPB1 to associate with sarcomeric proteins (Vicart et al. 1998). Mutations in HSPB5 are associated with a broad variety of neurological, cardiac, and muscular disorders. The R120G mutation results in an irregular protein structure and defective chaperone-like function (Bova et al. 1999), which may accelerate the accumulation of desmin aggregation, thereby leading to desmin-related myopathy and also early onset of cardiomyopathy (Selcen and Engel 2003; Vicart et al. 1998).
HSPB6 is abundantly expressed in skeletal muscle and heart in two complex formations: 43 kDa dimers and 470 kDa multimers. HSPB6 binds to itself and other HSPBs (HBPB1, HSPB5, and HSPB8) (Pipkin et al. 2003). Recently, HSPB6 overexpression was found to result in enhanced cardiac function by interacting with protein phosphatase 1, thereby inducing Ca2+ cycling and sarcoplasmic reticulum Ca2+ load (Qian et al. 2011). In addition, in HSPB6 transgenic rat ventricular cardiomyocytes, HSPB6 increases the phosphorylation at specific sites of the calcium regulatory protein phospholamban, via inhibition of protein phosphatase 1. As such, HSPB6 promotes the Ca2+ cycling in the sarcoplasmic reticulum and enhances the contractile function of the cardiomyocyte (Qian et al. 2011). Moreover, another study described HSPB6 to reduce the myocardial infarcted area, thereby conserving the heart integrity in mice with ischemia/reperfusion injury (Fan et al. 2006). Besides, the phosphorylation of HSPB6 at serine 16 was found to be required for attenuating ischemia/reperfusion-induced cell injury in mice, as the non-phosphorylatable HSPB6 induced apoptosis and necrosis, suppressed the autophagy activity, and subsequently depressed the cardiac functional recovery during ischemia and reperfusion (Qian et al. 2009).
HSPB7 is expressed in heart and skeletal muscle. In aged muscle, it was shown that both HSPB5 and HSPB7 expressions are dramatically increased (Doran et al. 2007). HSPB7 upregulation is also found in the muscular dystrophy-affected diaphragm, indicating that HSPB7 levels are induced under stress conditions. Furthermore, HSPB7 protects cells from protein aggregation, likely by facilitating cargo delivery to autophagosomes (Vos et al. 2010). Interestingly, HSPB4, HSPB6, or HSPB7 could not enhance the cellular capacity to chaperone heat-denatured luciferase, in contrast to HSPB1, indicating further functional differentiation of the HSPB members (Vos et al. 2010, 2011). In addition, co-localization of HSPB7 on myofibrils in cardiomyocytes is observed (Golenhofen et al. 2004), suggesting a protective role via conservation of the sarcomeric structure.
HSPB8 is strongly expressed in striated and smooth muscles, brain, and keratinocytes (Vos et al. 2008). Like HSPB1 and HSPB5, HSPB8 can also be phosphorylated in vitro. In contrast to HPB1 and HSPB5, phosphorylation of HSPB8 only marginally affects its tertiary and quaternary structure. Both wild-type and phosphorylated HSPB8 exist as low molecular mass oligomers. Unlike HSPB1 and HSPB5, where phosphorylation increases chaperone activity and reduces oligomeric size, phosphorylation of HSPB8 results in larger oligomeric structures and severely lowered chaperone activity (Basha et al. 2006). In in vitro experiments, HSPB8 interacts with several proteins and forms stoichiometric complexes with Bag3, a co-factor of HSPA1A (Carra et al. 2008a). The Bag3/HSPB8 complex was found to induce both translational arrest and autophagy, which may be beneficial in response to irreparable protein damage (Carra et al. 2008b, 2009). In addition, HSPB8 is cardioprotective in experimental models of myocardial ischemia. Overexpression of HSPB8 promotes cardiomyocyte survival after ischemia in mice (Depre et al. 2006) and attenuates the myocardial damage and contractile dysfunction in pig (Chen et al. 2011), whereas depletion of HSPB8 in mice with pressure overload contributes to the cardiac dysfunction and accelerates transition to heart failure (Qiu et al. 2011). Furthermore, studies show HSPB8 to conserve mitochondrial function and energy production, thereby attenuating oxidative stress in infarcted hearts (Marunouchi et al. 2014). In contrast to these beneficial effects of HSPB8 on cardiomyocyte function, overexpression of HSPB8 was also found to induce cardiac hypertrophy both in in vitro and in vivo model systems and reexpression of the cardiac fetal gene program and provoked cell growth pathways as well as proteasome activities (Depre et al. 2002; Hedhli et al. 2008). Therefore, the function of HSPB8 seems two-edged in heart diseases: HSPB8 reveals beneficial effects on myocardial ischemia by conserving the mitochondrial function and energy production, and HSPB8 is a mediator of cardiac hypertrophy and thereby results in heart failure.
Interestingly, various HSPB family members have common functions by translocating from cytoplasm to specific sarcomeric proteins upon different forms of stress (Table 2). During aging, HSPB1 was found to translocate from the cytoplasm of ventricular cardiomyocytes to sarcomeric actin in the Z line (Lutsch et al. 1997). Under acidic stress, HSPB1 translocates from the cytosol to the unfolded Ig domain of Titin on the I-band, to prevent its aggregation resulting in maintenance of titin function. Moreover, HSPB1 is co-localized with the titin spring in the elastic I-band region in dialated cardiomyopathy patients, while HSPB1 is mainly expressed in cytoplasm of cardiomyocytes in the healthy heart. In addition, phosphomimicking HSPB1 mutants did not alter its binding affinity to titin, compared to the wild-type HSPB1 (Kotter et al. 2014), indicating that binding of HSPB1 to titin is phosphorylation dependent. In the ischemic heart, co-localization of HSPB2 at the Z line of sarcomeres was enhanced (Yoshida et al. 1999). Moreover, in in vivo studies, HSPB5 was found to be soluble in the cytosol of cardiomyocytes under normal control conditions, while in the ischemic heart, HSPB5 was phosphorylated resulting in the transition of soluble HSPB5 to insoluble fractions and translocation from the cytosol to myofibrils. Furthermore, it was demonstrated in a pig model that HSPB5 strongly binds to titin after translocation to myofibrils. The chaperone activity is required to prevent the unfolding and irreversible derailment of myofibrils (Golenhofen et al. 1998, 1999, 2002). HSPB6 associates with HSPB5 and localizes in the distinct transverse bands in the similar pattern as sarcomeric actin, indicating that it probably modulates the contractile dynamics in cardiac myocytes through associating with sarcomeric actin (Pipkin et al. 2003).
Protective role of HSPB members in atrial fibrillation
So far, various HSPB members are found to be protective against AF (Table 3). In atrial tissue of patients with AF, HSPB1 localizes at sarcomeres (Brundel et al. 2006a). Furthermore, HSPB1 overexpression prevents the degradation of sarcomeric proteins in tachypaced HL-1 cardiomyocytes (Brundel et al. 2006a), indicating a prominent role for HSPB1 in conservation of the sarcomeric structure and function. Next to HSPB1, also HSPB6, HSPB7, and HSPB8 display protective effects against cardiomyocyte remodeling in tachypaced HL-1 cardiomyocytes (Ke et al. 2011). As several HSPB members can form hetero-oligomeric complexes with each other, the protective effect of the various members may be due to the supportive oligomeric structures with HSPB1 (van Montfort et al. 2001; Vos et al. 2008). As downregulation of endogenous HSPB1 did not impair the protective effects of HSPB6, HSPB7, and HSPB8 in tachypaced HL-1 cardiomyocytes, their effects seem independent of endogenous HSPB1 (Ke et al. 2011). Interestingly, all the protective HSPB members were able to reduce the formation of F-actin stress fibers, supporting the view that actin is the key target of the HSPB members in AF. Yet, the mode of action in preventing F-actin stress bundle formation of the four protective HSPB members seems to differ. Whereas HSPB8 interferes with the upstream tachypacing-induced RhoA GTPase activation, HSPB1, HSPB6, and HSPB7 do not. Rather, HSPB1, HSPB6, and HSPB7 bind to actin and directly inhibit G- to F-actin polymerization and/or stimulate depolymerization, indicating a protective role against tachycardia remodeling downstream of RhoA GTPase activation (Ke et al. 2011).
Next to the protective effects on F-actin stress bundle formation, HSPB1 conserves the calcium handling. HSPB1 overexpression protects against loss in Ca2+ transients and cell shortening in tachypaced HL-1 cardiomyocytes and this protective effect is phosphorylation-dependent, as a non-phosphorylatable HSPB1 mutant did not show an effect (Brundel et al. 2006a). In addition, the protective effect on the calcium handling may involve the direct modulation of ion channel function or modulation of specific kinases, resulting in the conservation of ion currents, including the L-type Ca2+ current (Christ et al. 2004). Previously, HSPs were found to regulate ion channel function in the heart and brain (Armstead and Hecker 2005; Ficker et al. 2003; Kashlan et al. 2007; Krieger et al. 2006). Some HSPs were found to interact directly with ion channels, such as HSPB5 with Na+ channels (Kashlan et al. 2007) and HSPA1A with cardiac K+ channel HERG (Ficker et al. 2003) and voltage-gated Ca2+ channels (Krieger et al. 2006), suggesting a possible role for HSPBs in AF attenuation by interacting with ion channels.
HSPBs may also protect against AF by affecting signaling cascades that are activated by AF. HSPB1 associates with several kinases, such as IkappaB kinase and c-Jun N-terminal kinase (JNK), thereby suppressing activation of the transcription factor NF-κB (Kammanadiminti and Chadee 2006; Park et al. 2003). Interestingly, these kinases were reported to be modulated during AF (Cardin et al. 2003; Li et al. 2001; Qi et al. 2008).
Finally, HSPBs may prevent cardiomyocyte remodeling via inhibition of proteases, such as calpain. In tachypaced Drosophila with dmHSP23 overexpression, likely the functional ortholog of human HSPB1 prevented the activation of calpain and myolysis and heart wall contractile dysfunction (Zhang et al. 2011). This finding is in line with a study showing that HSPB1 prevents ischemia/reperfusion-induced degradation of the contractile proteins cardiac troponin I and troponin T by interacting with the COOH-terminus and NH2-terminus, respectively. This interaction prevented calpain from cleaving cardiac troponin I and T and resulted in conservation of the contractile function in ventricular cardiomyocytes (Lu et al. 2008). Also, HSPB1 co-localizes with cardiac troponin T in ventricular cardiomyocytes after morphine withdrawal, thereby preventing its degradation by calpain and maintaining myocardial function (Martinez-Laorden et al. 2015). These findings together imply that HSPB1 binds to contractile proteins, thereby sequestering the proteolytic cleavage regions from calpain (Fig. 1).

AF induces a calcium overload in cardiomyocytes, which activates calcium-dependent neutral protease calpain. Calpain degrades contractile proteins and microtubule network resulting in structural remodeling, contractile dysfunction of cardiomyocytes, and AF progression. Elevated HSPB1 is found to inhibit calpain activity in tachypaced Drosophila. In addition, HSPB1 prevents degradation of cardiac troponins and may protect against depolymerization of α-tubulin by sequestering the proteolytic cleavage sites from calpain
HSPB in patients with AF
The experimental findings on the protective role of HSPB members in AF are in line with observations in paroxysmal and persistent AF patients. In patients with AF, an inverse correlation between the amount of HSPB1 expression and the level of myolysis and the duration of persistent AF is found (Brundel et al. 2006a). Patients with long-standing persistent AF reveal reduced levels of HSPB1 compared to persistent AF patients, suggesting that HSPB1 induction may represent a therapeutic target in long-standing persistent AF patients. Furthermore, HSPB members are found to represent a biomarker for AF onset and progression and may also predict the clinical outcome after interventions. A recent study showed that the serum HSPB1 levels of patients who received catheter ablation predict AF recurrences. Patients with high levels of HSPB1 in serum show improved maintenance rate of sinus rhythm (Hu et al. 2012). Because of the pleiotropic cardioprotective effects of HSPB1 on AF substrate formation, HSP inducers currently represent a class of drugs with promising therapeutic potential in clinical AF.
Therapeutic application of HSP induction in experimental and clinical AF
Previous research has demonstrated that the (genetic) induction of HSPB members provides prevention effect on tachycardia-induced structural remodeling and contractile dysfunction. A drug often used to boost HSP expression is geranylgeranylacetone (GGA) (Hoogstra-Berends et al. 2012). GGA is originally used as an anti-ulcer agent and is a non-toxic acyclic isoprenoid compound with a retinoid skeleton that induces HSP synthesis in various tissues, including gastric mucosa, intestine, liver, heart, retina, and the central nervous system (Katsuno et al. 2005; Ooie et al. 2001). GGA induces HSP expression probably via the activation of the heat shock transcription factor 1 (Ke et al. 2008). The protective effect of GGA-induced HSP expression on structural remodeling has been observed in experimental models of AF, suggesting that the induction of HSPs by GGA may have a potential value for clinical AF (Brundel et al. 2006a; Ke et al. 2008). In tachypaced Drosophila, GGA treatment protects against contractile dysfunction of the heart wall and structural remodeling (Zhang et al. 2011). Furthermore, in canine models for (acute) atrial ischemia-related AF and tachypacing-induced AF promotion, GGA treatment reveals protective effects against cardiomyocyte remodeling and consequently occurrence and recurrence of AF after cardioversion (Brundel et al. 2006a; Sakabe et al. 2008).
In addition to the pharmacological induction of HSPB, exercise is also found to induce HSPB levels and subsequently reveal cardioprotective effects. Various studies show that gene and protein levels of HSPB1 and HSPB6 are elevated after physical exercise in rat and mouse models. Interestingly, in these studies, HSPB1 and HSPB6 were phosphorylated, resulting in stabilization of myofilaments, restoration of disrupted contractile proteins, and consequently improved contractile function of the heart (Boluyt et al. 2006; Burniston 2009; Campos et al. 2012; de Moraes et al. 2015; Rinaldi et al. 2006; Sakamoto et al. 2006). Therefore, physical exercise may represent a promising therapeutic therapy to ameliorate the cardiac function and quality of life in patients with AF and maintain normal sinus rhythm after cardioversion, via induction of HSPB levels.
Conclusion
Various HSPB members conserve a healthy proteostasis of cardiomyocytes and thereby prevent AF onset and progression. Their mode of action is via the stabilization of the cardiomyocyte structure, thus conserving the contractile and electrophysiological function of the atria. Since compounds, such as GGA, and exercise are found to induce HSPB expression, these may represent promising novel therapeutic strategies to prevent AF onset and progression.
References
Armstead WM, Hecker JG (2005) Heat shock protein modulation of KATP and KCa channel cerebrovasodilation after brain injury. American Journal of Physiology - Heart and Circulatory Physiology 289(3):H1184–H1190. doi:10.1152/ajpheart.00276.2005
Ausma J, Wijffels M, Thone F, Wouters L, Allessie M, Borgers M (1997) Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation 96:3157–3163
Bakthisaran R, Tangirala R, Rao Ch M (2015) Small heat shock proteins: role in cellular functions and pathology. Biochim Biophys Acta 1854:291–319
Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science (New York, NY) 319:916–919
Basha E, Friedrich KL, Vierling E (2006) The N-terminal arm of small heat shock proteins is important for both chaperone activity and substrate specificity. J Biol Chem 281:39943–39952
Bennardini F, Wrzosek A, Chiesi M (1992) Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res 71(2):288–294
Boluyt MO, Brevick JL, Rogers DS, Randall MJ, Scalia AF, Li ZB (2006) Changes in the rat heart proteome induced by exercise training: increased abundance of heat shock protein hsp20. Proteomics 6:3154–3169
Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, Horwitz J (1999) Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A 96:6137–6142
Brundel BJ, Ausma J, van Gelder IC, Van der Want JJ, van Gilst WH, Crijns HJ, Henning RH (2002) Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc Res 54:380–389
Brundel BJ, Henning RH, Ke L, van Gelder IC, Crijns HJ, Kampinga HH (2006a) Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J Mol Cell Cardiol 41:555–562
Brundel BJ, Shiroshita-Takeshita A, Qi X, Yeh YH, Chartier D, van Gelder IC, Henning RH, Kampinga HH, Nattel S (2006b) Induction of heat shock response protects the heart against atrial fibrillation. Circ Res 99:1394–1402
Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, Ding L, Labeit S, Horwitz J, Leonard KR, Linke WA (2004) Association of the chaperone alphaB-crystallin with titin in heart muscle. J Biol Chem 279:7917–7924
Burniston JG (2009) Adaptation of the rat cardiac proteome in response to intensity-controlled endurance exercise. Proteomics 9:106–115
Campos JC, Queliconi BB, Dourado PM, Cunha TF, Zambelli VO, Bechara LR, Kowaltowski AJ, Brum PC, Mochly-Rosen D, Ferreira JC (2012) Exercise training restores cardiac protein quality control in heart failure. PLoS One 7:e52764
Cardin S, Li D, Thorin-Trescases N, Leung TK, Thorin E, Nattel S (2003) Evolution of the atrial fibrillation substrate in experimental congestive heart failure: angiotensin-dependent and -independent pathways. Cardiovasc Res 60:315–325
Carra S, Seguin SJ, Lambert H, Landry J (2008a) HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem 283:1437–1444
Carra S, Seguin SJ, Landry J (2008b) HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 4:237–239
Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH (2009) HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J Biol Chem 284:5523–5532
Chen L, Lizano P, Zhao X, Sui X, Dhar SK, Shen YT, Vatner DE, Vatner SF, Depre C (2011) Preemptive conditioning of the swine heart by H11 kinase/Hsp22 provides cardiac protection through inducible nitric oxide synthase. Am J Phys Heart Circ Phys 300:H1303–H1310
Chowdary TK, Raman B, Ramakrishna T, Rao CM (2004) Mammalian Hsp22 is a heat-inducible small heat-shock protein with chaperone-like activity. The Biochemical journal 381:379–387
Christ T, Boknik P, Wohrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, Dobrev D (2004) L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110:2651–2657
Cubedo J, Vilahur G, Casaní L, Mendieta G, Gómez-Jabalera E, Juan-Babot O, Padró T, Badimon L (2016) Targeting the molecular mechanisms of ischemic damage: Protective effects of alpha-crystallin-B. Int J Cardiol 215:406–416. doi:10.1016/j.ijcard.2016.04.072
Depre C, Hase M, Gaussin V, Zajac A, Wang L, Hittinger L, Ghaleh B, Yu X, Kudej RK, Wagner T, Sadoshima J, Vatner SF (2002) H11 kinase is a novel mediator of myocardial hypertrophy in vivo. Circ Res 91:1007–1014
de Groot NM, Houben RP, Smeets JL, Boersma E, Schotten U, Schalij MJ, Crijns H, Allessie MA (2010) Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: epicardial breakthrough. Circulation 122:1674–1682
Depre C, Wang L, Sui X, Qiu H, Hong C, Hedhli N, Ginion A, Shah A, Pelat M, Bertrand L, Wagner T, Gaussin V, Vatner SF (2006) H11 kinase prevents myocardial infarction by preemptive preconditioning of the heart. Circ Res 98:280–288
Dobrev D, Carlsson L, Nattel S (2012) Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov 11:275–291
Doran P, Gannon J, O'Connell K, Ohlendieck K (2007) Aging skeletal muscle shows a drastic increase in the small heat shock proteins alphaB-crystallin/HspB5 and cvHsp/HspB7. Eur J Cell Biol 86:629–640
Fan GC, Kranias EG (2011) Small heat shock protein 20 (HspB6) in cardiac hypertrophy and failure. J Mol Cell Cardiol 51(4):574–577. doi:10.1016/j.yjmcc.2010.09.013
Fan GC, Yuan Q, Song G, Wang Y, Chen G, Qian J, Zhou X, Lee YJ, Ashraf M, Kranias EG (2006) Small heat-shock protein Hsp20 attenuates beta-agonist-mediated cardiac remodeling through apoptosis signal-regulating kinase 1. Circ Res 99:1233–1242
Fan GC, Ren X, Qian J, Yuan Q, Nicolaou P, Wang Y, Jones WK, Chu G, Kranias EG (2015) Novel Cardioprotective Role of a Small Heat-Shock Protein, Hsp20, Against Ischemia/Reperfusion Injury. Circulation 111:1792–1799. doi:10.1161/01.CIR.0000160851.41872
Ficker E, Dennis AT, Wang L, Brown AM (2003) Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res 92:e87–100
Ghosh JG, Houck SA, Clark JI (2007) Interactive domains in the molecular chaperone human alphaB crystallin modulate microtubule assembly and disassembly. PLoS One 2:e498
Golenhofen N, Ness W, Koob R, Htun P, Schaper W, Drenckhahn D (1998) Ischemia-induced phosphorylation and translocation of stress protein alpha B-crystallin to Z lines of myocardium. Am J Phys 274:H1457–H1464
Golenhofen N, Htun P, Ness W, Koob R, Schaper W, Drenckhahn D (1999) Binding of the stress protein alpha B-crystallin to cardiac myofibrils correlates with the degree of myocardial damage during ischemia/reperfusion in vivo. J Mol Cell Cardiol 31:569–580
Golenhofen N, Arbeiter A, Koob R, Drenckhahn D (2002) Ischemia-induced association of the stress protein alpha B-crystallin with I-band portion of cardiac titin. J Mol Cell Cardiol 34:309–319
Golenhofen N, Perng MD, Quinlan RA, Drenckhahn D (2004) Comparison of the small heat shock proteins alphaB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem Cell Biol 122:415–425
Golenhofen N, Redel A, Wawrousek EF, Drenckhahn D (2006) Ischemia-induced increase of stiffness of alphaB-crystallin/HSPB2-deficient myocardium. Pflugers Archiv: European journal of physiology 451:518–525
Grose JH, Langston K, Wang X, Squires S, Mustafi SB, Hayes W, Neubert J, Fischer SK, Fasano M, Saunders GM, Dai Q, Christians E, Lewandowski ED, Ping P, Benjamin IJ (2015) Characterization of the cardiac overexpression of HSPB2 reveals mitochondrial and myogenic roles supported by a cardiac HspB2 interactome. PLoS One 10:e0133994
Hedhli N, Wang L, Wang Q, Rashed E, Tian Y, Sui X, Madura K, Depre C (2008) Proteasome activation during cardiac hypertrophy by the chaperone H11 kinase/Hsp22. Cardiovasc Res 77:497–505
Hoogstra-Berends F, Meijering RA, Zhang D, Heeres A, Loen L, Seerden JP, Kuipers I, Kampinga HH, Henning RH, Brundel BJ (2012) Heat shock protein-inducing compounds as therapeutics to restore proteostasis in atrial fibrillation. Trends in cardiovascular medicine 22:62–68
Hu YF, Yeh HI, Tsao HM, Tai CT, Lin YJ, Chang SL, Lo LW, Tuan TC, Suenari K, Li CH, Chao TF, Chen SA (2012) Electrophysiological correlation and prognostic impact of heat shock protein 27 in atrial fibrillation. Circulation Arrhythmia and electrophysiology 5:334–340
Ishiwata T, Orosz A, Wang X, Mustafi SB, Pratt GW, Christians ES, Boudina S, Abel ED, Benjamin IJ (2012) HSPB2 is dispensable for the cardiac hypertrophic response but reduces mitochondrial energetics following pressure overload in mice. PLoS One 7:e42118
Islamovic E et al (2007) Importance of Small Heat Shock Protein 20 (Hsp20) C-Terminal Extension in Cardioprotection. J Mol Cell Cardiol 42(4):862–869
Juo LY, Liao WC, Shih YL, Yang BY, Liu AB, Yan YT (2016) HSPB7 interacts with dimerized FLNC and its absence results in progressive myopathy in skeletal muscles. J Cell Sci 129(8):1661–1670. doi:10.1242/jcs.179887
Kadono T, Zhang XQ, Srinivasan S, Ishida H, Barry WH, Benjamin IJ (2006) CRYAB and HSPB2 deficiency increases myocyte mitochondrial permeability transition and mitochondrial calcium uptake. J Mol Cell Cardiol 40:783–789
Kammanadiminti SJ, Chadee K (2006) Suppression of NF-kappaB activation by Entamoeba histolytica in intestinal epithelial cells is mediated by heat shock protein 27. J Biol Chem 281:26112–26120
Kampinga HH, Bergink S (2016) Heat shock proteins as potential targets for protective strategies in neurodegeneration. The Lancet Neurology 15:748–759
Kashlan OB, Mueller GM, Qamar MZ, Poland PA, Ahner A, Rubenstein RC, Hughey RP, Brodsky JL, Kleyman TR (2007) Small heat shock protein alphaA-crystallin regulates epithelial sodium channel expression. J Biol Chem 282:28149–28156
Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G (2005) Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A 102:16801–16806
Kawano F, Fujita R, Nakai N, Terada M, Ohira T, Ohira Y (2012) HSP25 can modulate myofibrillar desmin cytoskeleton following the phosphorylation at Ser15 in rat soleus muscle. J Appl Physiol (1985) 112(1):176–186. doi:10.1152/japplphysiol.00783.2011
Ke L, Qi XY, Dijkhuis AJ, Chartier D, Nattel S, Henning RH, Kampinga HH, Brundel BJ (2008) Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J Mol Cell Cardiol 45:685–693
Ke L, Meijering RA, Hoogstra-Berends F, Mackovicova K, Vos MJ, Van Gelder IC, Henning RH, Kampinga HH, Brundel BJ (2011) HSPB1, HSPB6, HSPB7 and HSPB8 protect against RhoA GTPase-induced remodeling in tachypaced atrial myocytes. PLoS One 6:e20395
Kirmanoglou K, Hannekum A, Schafler AE (2004) Expression of mortalin in patients with chronic atrial fibrillation. Basic Res Cardiol 99:404–408
Kirubakaran S, Chowdhury RA, Hall MC, Patel PM, Garratt CJ, Peters NS (2015) Fractionation of electrograms is caused by colocalized conduction block and connexin disorganization in the absence of fibrosis as AF becomes persistent in the goat model. Heart rhythm: the official journal of the Heart Rhythm Society 12:397–408
Kotter S, Unger A, Hamdani N, Lang P, Vorgerd M, Nagel-Steger L, Linke WA (2014) Human myocytes are protected from titin aggregation-induced stiffening by small heat shock proteins. J Cell Biol 204:187–202
Krief S, Faivre JF, Robert P, Le Douarin B, Brument-Larignon N, Lefrère I, Bouzyk MM, Anderson KM, Greller LD, Tobin FL, Souchet M, Bril A (1999) Identification and characterization of cvHsp. A novel human small stress protein selectively expressed in cardiovascular and insulin-sensitive tissues. J Biol Chem 274(51):36592–600
Krieger A, Radhakrishnan K, Pereverzev A, Siapich SA, Banat M, Kamp MA, Leroy J, Klockner U, Hescheler J, Weiergraber M, Schneider T (2006) The molecular chaperone hsp70 interacts with the cytosolic II-III loop of the Cav2.3 E-type voltage-gated Ca2+ channel. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology 17:97–110
Landry J, Huot J (1995) Modulation of actin dynamics during stress and physiological stimulation by a signaling pathway involving p38 MAP kinase and heat-shock protein 27. Biochemistry and cell biology = Biochimie et biologie cellulaire 73:703–707
Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J (1995) Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol 15:505–516
Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S (2001) Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation 104:2608–2614
Liu L, Zhang X, Qian B, Min X, Gao X, Li C, Cheng Y, Huang J (2007) Over-expression of heat shock protein 27 attenuates doxorubicin-induced cardiac dysfunction in mice. Eur J Heart Fail 9(8):762–9
Lu XY, Chen L, Cai XL, Yang HT (2008) Overexpression of heat shock protein 27 protects against ischaemia/reperfusion-induced cardiac dysfunction via stabilization of troponin I and T. Cardiovasc Res 79:500–508
Lutsch G, Vetter R, Offhauss U, Wieske M, Grone HJ, Klemenz R, Schimke I, Stahl J, Benndorf R (1997) Abundance and location of the small heat shock proteins HSP25 and alphaB-crystallin in rat and human heart. Circulation 96:3466–3476
Mandal K, Torsney E, Poloniecki J, Camm AJ, Xu Q, Jahangiri M (2005) Association of high intracellular, but not serum, heat shock protein 70 with postoperative atrial fibrillation. Ann Thorac Surg 79:865–871 discussion 871
Martinez-Laorden E, Almela P, Milanes MV, Laorden ML (2015) Expression of heat shock protein 27 and troponin T and troponin I after naloxone-precipitated morphine withdrawal. Eur J Pharmacol 766:142–150
Marunouchi T, Inomata S, Sanbe A, Takagi N, Tanonaka K (2014) Protective effect of geranylgeranylacetone via enhanced induction of HSPB1 and HSPB8 in mitochondria of the failing heart following myocardial infarction in rats. Eur J Pharmacol 730:140–147
Mogk A, Schlieker C, Friedrich KL, Schonfeld HJ, Vierling E, Bukau B (2003) Refolding of substrates bound to small Hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J Biol Chem 278:31033–31042
van Montfort RL, Basha E, Friedrich KL, Slingsby C, Vierling E (2001) Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol 8:1025–1030
de Moraes WM, Melara TP, de Souza PR, Guimaraes Fde S, Bozi LH, Brum PC, Medeiros A (2015) Impact of leucine supplementation on exercise training induced anti-cardiac remodeling effect in heart failure mice. Nutrients 7:3751–3766
Nakagawa M, Tsujimoto N, Nakagawa H, Iwaki T, Fukumaki Y, Iwaki A (2001) Association of HSPB2, a member of the small heat shock protein family, with mitochondria. Exp Cell Res 271:161–168
Nicolaou P, Knöll R, Haghighi K, Fan GC, Dorn GW 2nd, Hasenfub G, Kranias EG (2008) Human mutation in the anti-apoptotic heat shock protein 20 abrogates its cardioprotective effects. J Biol Chem 283(48):33465–33471. doi:10.1074/jbc.M802307200
Ooie T, Takahashi N, Saikawa T, Nawata T, Arikawa M, Yamanaka K, Hara M, Shimada T, Sakata T (2001) Single oral dose of geranylgeranylacetone induces heat-shock protein 72 and renders protection against ischemia/reperfusion injury in rat heart. Circulation 104:1837–1843
Park KJ, Gaynor RB, Kwak YT (2003) Heat shock protein 27 association with the I kappa B kinase complex regulates tumor necrosis factor alpha-induced NF-kappa B activation. J Biol Chem 278:35272–35278
Paulsen G, Lauritzen F, Bayer ML, Kalhovde JM, Ugelstad I, Owe SG, Hallén J, Bergersen LH, Raastad T (2009) Subcellular movement and expression of HSP27, alphaB-crystallin, and HSP70 after two bouts of eccentric exercise in humans. J Appl Physiol (1985) 107(2):570–582. doi:10.1152/japplphysiol.00209.2009
Perng MD, Cairns L, van den IJssel P, Prescott A, Hutcheson AM, Quinlan RA (1999) Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci 112(Pt 13):2099–2112
Pipkin W, Johnson JA, Creazzo TL, Burch J, Komalavilas P, Brophy C (2003) Localization, macromolecular associations, and function of the small heat shock-related protein HSP20 in rat heart. Circulation 107:469–476
Qi XY, Yeh YH, Xiao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJ, Dobrev D, Nattel S (2008) Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res 103:845–854
Qian J, Ren X, Wang X, Zhang P, Jones WK, Molkentin JD, Fan GC, Kranias EG (2009) Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ Res 105:1223–1231
Qian J, Vafiadaki E, Florea SM, Singh VP, Song W, Lam CK, Wang Y, Yuan Q, Pritchard TJ, Cai W, Haghighi K, Rodriguez P, Wang HS, Sanoudou D, Fan GC, Kranias EG (2011) Small heat shock protein 20 interacts with protein phosphatase-1 and enhances sarcoplasmic reticulum calcium cycling. Circ Res 108:1429–1438
Qiu H, Lizano P, Laure L, Sui X, Rashed E, Park JY, Hong C, Gao S, Holle E, Morin D, Dhar SK, Wagner T, Berdeaux A, Tian B, Vatner SF, Depre C (2011) H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation 124:406–415
Rinaldi B, Corbi G, Boccuti S, Filippelli W, Rengo G, Leosco D, Rossi F, Filippelli A, Ferrara N (2006) Exercise training affects age-induced changes in SOD and heat shock protein expression in rat heart. Exp Gerontol 41:764–770
Sakabe M, Shiroshita-Takeshita A, Maguy A, Brundel BJ, Fujiki A, Inoue H, Nattel S (2008) Effects of a heat shock protein inducer on the atrial fibrillation substrate caused by acute atrial ischaemia. Cardiovasc Res 78:63–70
Sakamoto M, Minamino T, Toko H, Kayama Y, Zou Y, Sano M, Takaki E, Aoyagi T, Tojo K, Tajima N, Nakai A, Aburatani H, Komuro I (2006) Upregulation of heat shock transcription factor 1 plays a critical role in adaptive cardiac hypertrophy. Circ Res 99:1411–1418
Sanbe A, Yamauchi J, Miyamoto Y, Fujiwara Y, Murabe M, Tanoue A (2007) Interruption of CryAB-amyloid oligomer formation by HSP22. J Biol Chem 282:555–563
Schafler AE, Kirmanoglou K, Pecher P, Hannekum A, Schumacher B (2002) Overexpression of heat shock protein 60/10 in myocardium of patients with chronic atrial fibrillation. Ann Thorac Surg 74:767–770
Selcen D, Engel AG (2003) Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann Neurol 54:804–810
St Rammos K, Koullias GJ, Hassan MO, Argyrakis NP, Voucharas CG, Scarupa SJ, Cowte TG (2002) Low preoperative HSP70 atrial myocardial levels correlate significantly with high incidence of postoperative atrial fibrillation after cardiac surgery. Cardiovascular surgery (London, England) 10:228–232
Sugiyama Y, Suzuki A, Kishikawa M, Akutsu R, Hirose T, Waye MM, Tsui SK, Yoshida S, Ohno S (2000) Muscle develops a specific form of small heat shock protein complex composed of MKBP/HSPB2 and HSPB3 during myogenic differentiation. J Biol Chem 275:1095–1104
Suzuki A, Sugiyama Y, Hayashi Y, Nyu-i N, Yoshida M, Nonaka I, Ishiura S, Arahata K, Ohno S (1998) MKBP, a novel member of the small heat shock protein family, binds and activates the myotonic dystrophy protein kinase. J Cell Biol 140:1113–1124
Todd DM, Fynn SP, Walden AP, Hobbs WJ, Arya S, Garratt CJ (2004) Repetitive 4-week periods of atrial electrical remodeling promote stability of atrial fibrillation: time course of a second factor involved in the self-perpetuation of atrial fibrillation. Circulation 109:1434–1439
Veinger L, Diamant S, Buchner J, Goloubinoff P (1998) The small heat-shock protein IbpB from Escherichia coli stabilizes stress-denatured proteins for subsequent refolding by a multichaperone network. J Biol Chem 273:11032–11037
van de Klundert FA, Gijsen ML, van den IPR, Snoeckx LH, de Jong WW (1998) Alpha B-crystallin and hsp25 in neonatal cardiac cells—differences in cellular localization under stress conditions. Eur J Cell Biol 75:38–45
Vander Heide RS (2002) Increased expression of HSP27 protects canine myocytes from simulated ischemia-reperfusion injury. American Journal of Physiology - Heart and Circulatory Physiology 282(3):H935–H941. doi:10.1152/ajpheart.00660.2001
Verschuure P, Tatard C, Boelens WC, Grongnet JF, David JC (2003) Expression of small heat shock proteins HspB2, HspB8, Hsp20 and cvHsp in different tissues of the perinatal developing pig. Eur J Cell Biol 82:523–530
Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, Chateau D, Chapon F, Tome F, Dupret JM, Paulin D, Fardeau M (1998) A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet 20:92–95
Vos MJ, Hageman J, Carra S, Kampinga HH (2008) Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry 47:7001–7011
Vos MJ, Kanon B Fau-Kampinga HH, Kampinga HH (2009) HSPB7 is a SC35 speckle resident small heat shock protein. Biochim Biophys Acta 1793(8):1343–1353. doi:10.1016/j.bbamcr.2009.05.005
Vos MJ, Zijlstra MP, Kanon B, van Waarde-Verhagen MA, Brunt ER, Oosterveld-Hut HM, Carra S, Sibon OC, Kampinga HH (2010) HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum Mol Genet 19:4677–4693
Vos MJ, Zijlstra MP, Carra S, Sibon OC, Kampinga HH (2011) Small heat shock proteins, protein degradation and protein aggregation diseases. Autophagy 7:101–103
Westerheide SD, Morimoto RI (2005) Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem 280:33097–33100
Yoshida K, Aki T, Harada K, Shama KM, Kamoda Y, Suzuki A, Ohno S (1999) Translocation of HSP27 and MKBP in ischemic heart. Cell Struct Funct 24:181–185
Zhang D, Ke L, Mackovicova K, Van Der Want JJ, Sibon OC, Tanguay RM, Morrow G, Henning RH, Kampinga HH, Brundel BJ (2011) Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for atrial fibrillation. J Mol Cell Cardiol 51:381–389
Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra-Berends F, Tadevosyan A, Cubukcuoglu Deniz G, Durdu S, Akar AR, Sibon OC, Nattel S, Henning RH, Brundel BJ (2014) Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 129:346–358
Acknowledgements
We acknowledge the support from The Netherlands Cardiovascular Research Initiative and Dutch Heart Foundation CVON2014-40 DOSIS and CVON-STW2016-14728 AFFIP, Dutch Heart Foundation (2013T096 and 2013T144), and LSH-TKI (40-43100-98-008).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hu, X., Van Marion, D.M.S., Wiersma, M. et al. The protective role of small heat shock proteins in cardiac diseases: key role in atrial fibrillation. Cell Stress and Chaperones 22, 665–674 (2017). https://doi.org/10.1007/s12192-017-0799-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-017-0799-4