
Abstract
By environmental stresses, cells can initiate a signaling pathway in which eukaryotic translation initiation factor 2-alpha (eIF2-α) is involved to regulate the response. Phosphorylation of eIF2-α results in the reduction of overall protein neogenesis, which allows cells to conserve resources and to reprogram energy usage for effective stress control. To investigate the role of eIF2-α in cell stress responses, we conducted a viability-based compound screen under endoplasmic reticulum (ER) stress condition, and identified 1-(4-biphenylylcarbonyl)-4-(5-bromo-2-methoxybenzyl) piperazine oxalate (AMC-01) and its derivatives as eIF2-α-inactivating chemical. Molecular characterization of this signaling pathway revealed that AMC-01 induced inactivation of eIF2-α by phosphorylating serine residue 51 in a dose- and time-dependent manner, while the negative control compounds did not affect eIF2-α phosphorylation. In contrast with ER stress induction by thapsigargin, phosphorylation of eIF2-α persisted for the duration of incubation with AMC-01. By pathway analysis, AMC-01 clearly induced the activation of protein kinase RNA-activated (PKR) kinase and nuclear factor-κB (NF-κB), whereas it did not modulate the activity of PERK or heme-regulated inhibitor (HRI). Finally, we could detect a lower protein translation rate in cells incubated with AMC-01, establishing AMC-01 as a potent chemical probe that can regulate eIF2-α activity. We suggest from these data that AMC-01 and its derivative compounds can be used as chemical probes in future studies of the role of eIF2-α in protein synthesis-related cell physiology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Various types of stress, such as nutrient excess or deprivation, xenobiotics, or DNA damage, can cause cellular injury and disruption of cellular processes, including translation (Holcik and Sonenberg 2005; Sonenberg and Hinnebusch 2009). In this respect, protein synthesis is among the most critical cellular pathways for maintaining homeostasis, and cells carefully control translational regulation to regulate gene expression and relieve cell stress. Accordingly, this process plays a crucial role in cell growth and proliferation, and also contributes to pathophysiology (Silvera et al. 2010).
Protein translation can be controlled at each of the three major steps: initiation, elongation, and termination. A critical event in the initiation process is phosphorylation of the alpha (α) subunit of protein translation initiation factor eIF2 (eIF2-α) at serine residue 51, a modification that blocks initiation (Koromilas 2015) that can be mediated by kinases that respond to diverse forms of stress (Wang and Kaufman 2014). Family members among the serine-threonine kinases known as eIF2-α kinases are responsible for the phosphorylation of eIF2-α (Donnelly et al. 2013). This kinase family includes heme-regulated inhibitor (HRI), general control non-depressible-2 (GCN2), interferon-inducible protein kinase RNA-activated (PKR), and PKR-like endoplasmic reticulum-resident kinase (PERK). HRI becomes activated when cells are insufficient in iron or heme or are exposed to oxidative stress (Han et al. 2001). GCN2 can be activated by uncharged transfer RNA (tRNA) in response to amino acid starvation, which results in the induction of amino acid biosynthetic genes (Deval et al. 2009). The activity of PERK is induced by the presence of unfolded proteins in the endoplasmic reticulum (ER) and results in a reduction in protein synthesis that can prevent the accumulation of incorrectly folded or unfolded proteins (Healy et al. 2009; Szegezdi et al. 2006). Finally, PKR is activated by double-stranded (ds) RNA produced during virus replication, which results in the inhibition of viral and host protein synthesis (Galluzzi et al. 2008; Ito et al. 1999; Nakamura et al. 2010; Onuki et al. 2004; Ruvolo et al. 2001). Each of these enzymes exhibits many sequence similarities, particularly in the protein kinase domain (KD) (Harding et al. 1999).
Because eIF2-α is very tightly regulated, dysregulation of eIF2-α can lead to pathological conditions (Balachandran and Barber 2004) and it is positively accepted that phosphorylation-dependent temporal inactivation of eIF2-α can make cells resistant to various stress-related cell death conditions, including ER stress-induced cell death. By contrast, the role of sustained eIF2-α phosphorylation in final cell fate decisions remains unclear. In this context, one small molecule, salubrinal, can sustain eIF2-α phosphorylation and shows cytoprotective properties under ER stress induction conditions (Boyce et al. 2005). However, salubrinal has a relatively high range of working concentrations and its molecular target profile remains unclear, suggesting that the identification of a new chemical probe would be useful.
To identify a new chemical probe for investigating eIF2-α function, we performed high throughput compound screening, isolated candidate compounds, and characterized AMC-01 and its derivative compounds, which were enhanced using a combined assessment of both chemical properties and biological effects. We suggest from our findings that AMC-01 and its derivative compounds can be used as potent chemical probes to induce eIF2-α inactivation in future studies of protein translation in various biomedical contexts.
Materials and methods
Cell culture
CSM 14.1 cells, a rat immortalized mesencephalic neural progenitor cell line, were maintained in DMEM supplemented with 10 % fetal bovine serum (FBS), 100 μg/ml streptomycin, 100 IU penicillin, and 1 % L-glutamine. MCF-7 cells, a human breast carcinoma cell line, were maintained in RPMI-1640 with 10 % FBS, 100 μg/ml streptomycin, 100 IU penicillin, and 1 % L-glutamine.
Compound screening
The high throughput screening efforts to isolate inhibitors of ER stress-induced cell death were described in a previous report (Kim et al. 2009).
Cell viability assay
Cell viability was evaluated using an assay to measure intracellular ATP content (CellTiterGlo; Promega, Madison, WI) according to the manufacturer’s protocol with some modifications. After a 1 h pre-incubation with compounds, cells were treated with cell death-inducing agents for 24 h. Each well contained 10-μl assay solution, and raw luminescence data were analyzed using GraphPad Prism (GraphPad, La Jolla, CA) to evaluate relative cell survival rate. Raw values from untreated wells were adjusted for an indication of 100 % relative survival.
Chemicals and reagents
All chemical compounds isolated in the high throughput screen were purchased from Chembridge (San Diego, CA). The IDs of probe compounds in the “hit2 lead.com” site are as follows: AMC-01 (5990041), AMC-02 (5955734), AMC-03 (6035098), and AMC-04 (5990137). The ID numbers of negative control compounds for mechanistic studies and structure-activity relationship analyses are 6033233 (inactive analog: IA-1), 5397316 (IA-2), 6033487 (IA-3), 5990076 (IA-4), 5935965 (IA-5), 5951294 (IA-6), 5996087 (IA-7), 6036165 (IA-8), 5952061 (IA-9), and 5998646 (AMC-05). The ID number for the positive control (salubrinal) was 5147990. The ER stress inducer thapsigargin was from Enzo Life Science (Farmingdale, NY). Tunicamycin, VP16, staurosporine, and all other chemicals for experiments were purchased from Sigma-Aldrich (St. Louis, MO).
Western blotting analysis
Cells were extracted by scraping in the presence of lysis buffer (50 mM Tris–HCl pH 7.4, 1 % IGEPAL, 200 mM NaCl, 1 mM MgCl2, 1 mM sodium orthovanadate, 2.5 mM β-glycerophosphate, 1 mM sodium fluoride, and a protease inhibitor cocktail from Roche (Basel, Switzerland)). After extraction, 30 μg total cell protein extract was resolved in a sodium dodecyl sulfate-polyacrylamide gel by electrophoresis (SDS-PAGE). Separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P membrane; Millipore, Billerica, MA). After blocking, membranes were incubated with primary antibodies at a 1:1000 dilution, and with secondary antibodies at a 1:5000 dilution. After washing, the protein bands were visualized using horseradish peroxidase-conjugated immunoglobulin G (Bio-Rad and Cell Signaling) with a chemiluminescent substrate (SuperSignal West Pico; Pierce, Rockford, IL). As a loading control, membranes were stripped and re-probed with an anti-α-tubulin antibody.
Antibodies used in this study were as follows: anti-caspase-3 (Cell Signaling Technology; catalog #9665), anti-poly-ADP ribosyl polymerase (Cell Signaling Technology; #9532), anti-phospho-eIF2-α (Cell Signaling Technology; #3597), anti-eIF2-α (Santa Cruz; #sc-11386), anti-phospho-c-Jun (Cell Signaling Technology; #9164), anti-c-Jun (Santa Cruz; #sc-1694), anti-phospho-p38 MAPK (Cell Signaling Technology; #9216), anti-p38 MAPK (Santa Cruz; #sc-728), anti-phospho-PERK (Santa Cruz; #32577), anti-PERK (Cell Signaling Technology; #5683), anti-phospho-PKR (Life Technologies; #44-668G), anti-PKR (Cell Signaling Technology; #3072), anti-phospho-GCN2 (Abcam; #75836), anti-GCN2 (Cell Signaling Technology; #3302), anti-phospho-p65 (Cell Signaling Technology; #3031), anti-p65 (Cell Signaling Technology; #4764), anti-β-actin (Novus Biologicals; #NB 600–501), anti α-tubulin (Sigma; cat#T9026), anti ATF4 (Santa cruz; cat#sc-200), anti Cyclin D1 (Cell signaling technology; cat#2978), anti ATF6 (Abcam; cat#65838), anti IRE1-α (Abcam; cat#37073), and anti-phospho IRE1-α (Abcam; cat#48187).
XBP-1 splicing assay
X-box binding protein 1 (XBP-1) splicing in response to stress induction was examined by reverse transcription-polymerase chain reaction (RT-PCR) using total RNA extracted from cells. CSM 14.1 cells were cultured with 25 μM AMC-01 in DMSO for 1 h, followed by treatment with TG for 2 h. Total RNA samples (1 μg) were analyzed by RT-PCR using the following primer set for rat XBP-1: forward, 5′-CAGAGTAGCAGCACCAGACTGC-3′; reverse, 5′-TCCTTCTGGGTAGACCTCTGGGAG-3′.
Phospho-eIF2-α ELISA
Enzyme-linked immune sorbent assay (ELISA) was used to quantitatively evaluate the ratio of phospho-eIF2-α to total eIF2-α with a “PathScan Sandwich ELISA kit” to detect phospho-eIF2-α (Ser51) and total eIF2-α (Cell Signaling Technology; #7286 and #7295) according to the manufacturer’s protocol. Briefly, after incubation with compounds, cells were lyzed with lysis buffer (supplied by manufacturer), and the protein concentrations were calculated using the bicinchoninic acid (BCA assay). A total of 30 μg protein extract was used for measurements. The eIF2-α induction rate was evaluated by normalizing the raw value of phospho-eIF2-α to that of total eIF2-α. The calculated value of p-eIF2-α/eIF2-α from DMSO-treated samples was adjusted to 1, and the relative fold-induction of the ratio was recorded.
Evaluation of the cap-dependent translation rate
The cap-dependent translation rate was evaluated by calculating the rate of reporter gene translation using a pRMF reporter vector (a kind gift from Dr. Sung Key Jang, Pohang University of Science and Technology, Pohang, Republic of Korea). After plating in 6-well plates, MCF-7 cells were transfected with pRMF vector (1 μg) for 24 h. After incubation with compounds, cell lysates were extracted in cell extract buffer. An equal amount of lysate was subjected to analysis using a dual-luciferase assay to calculate the cap-dependent translation of renilla reporter and internal ribosomal entry site (IRES)-dependent translation of the firefly reporter gene. Briefly, raw values of the renilla reporter were normalized to the raw value of the firefly reporter in one sample, as reported previously (Chappell et al. 2000; Ling et al. 2005). Each experiment was conducted twice and three independent samples were analyzed in each experiment.
Results
Identification of small molecule inhibitors of ER stress-induced cell death
Prolonged stress induced by ER dysfunction ultimately results in cell death, usually with an apoptotic phenotype (Schroder and Kaufman 2005; Tabas and Ron 2011). To identify chemical probes of ER stress-induced cell death, we performed high-throughput screening and isolated small molecule inhibitors of thapsigargin-induced cell death using CSM14.1, a rat neuroprogenitor cell line (Kim et al. 2009). As previously reported, 198 primary hit compounds had been identified as inhibitors of ER stress-induced cell death in a screen of a commercially available chemical library of 50,000 compounds (ChemBridge Corp., San Diego, CA) (Kim et al. 2009). A counter-screen of primary hit compounds revealed that 26 were cytoprotective for undifferentiated CSM14.1 cells against thapsigargin- or tunicamycin-induced cell death. Based on a chemical classification analysis, we found that 4 of 26 hit compounds shared a benzyl piperazine benzamide core structure (Fig. 1a). Figure 1b shows raw data of four plates acquired from the primary screen in which compounds AMC-01~04 were identified as strong inhibitors of thapsigargin-induced cell death in CSM14.1 cells.

Identification of benzyl piperazine benzamide (AMC) compounds as inhibitors of ER stress-induced cell death. a Structures of the AMC compounds with a benzyl piperazine benzamide scaffold. Inactive analog compounds are indicated as IA-1 and IA-2. b Raw data pictograph of plates for a high-throughput screen in which AMC-01~04 were isolated as hit compounds. Relative ATP contents were interpreted as a “relative survival value” (y-axis) that was plotted against the well number in a 96-well plate (A1 to H12). Wells A1 to D1 represent positive controls (salubrinal 100 μM + thapsigargin 15 μM), wells E1 to H1 are negative controls (DMSO + thapsigargin 15 μM), and wells A12 to H12 are assay controls (DMSO only) that imply a 100 % survival rate. c Four benzyl piperazine benzamide compounds and salubrinal were compared with respect to cytoprotective activity against ER stress (thapsigargin)-induced death in CSM14.1 cells. Relative survival data were evaluated using an ATP content assay (upper graph) or flow cytometry analysis of cells stained with Annexin V-FITC and propidium iodide (lower graph). Data are expressed as percentages of control cells with DMSO without thapsigargin (upper graph) or as percentages of Annexin V-negative cells (lower graph). As a positive control, cells were treated with 100 μM Sal (salubrinal). The final concentrations of AMC compounds were 25 μM (n = 2, data represent the means ± SD). d ER stress-induced apoptotic caspase-3 activation was inhibited by AMC-01. Cells were pre-incubated with DMSO or AMC-01 and then treated with thapsigargin for 12 h. Cell lysates were harvested and used for western blotting to assess the cleavage of caspase-3 and poly ADP-ribosyl polymerase (PARP). e CSM14.1 cells were treated with thapsigargin (15 μM) and increasing concentrations of AMC compounds (AMC-01 to AMC-04), inactive analogs (IA-1 and IA-2), or salubrinal (Sal). ATP levels in cells were measured in triplicate, and were interpreted as relative survival normalized to DMSO controls. A dose–response curve for each compound is shown and the data in the graph represent the means ± S.D. The calculated EC50 values are presented in Table 1
To confirm the protective effects of these four structurally related compounds, fresh stocks of each compound were obtained and subjected to an additional round of cell death analysis. Both the cell proliferation assay (Fig. 1c, upper) and flow cytometry analysis (Fig. 1c, lower) revealed that all four compounds tested were cytoprotective against thapsigargin-induced cell death in CSM14.1 cells. The AMC compounds exhibited cytoprotective efficiency at 25 μM that was comparable to 100 μM salubrinal, a well-characterized inhibitor of ER stress-induced cell death (Boyce et al. 2005). Pre-incubation of cells with AMC-01 clearly inhibited thapsigargin-induced caspase-3 activation and the cleavage of poly ADP-ribosyl polymerase (PARP), implying that apoptotic cell death pathways were suppressed by AMC-01 (Fig. 1d).
To compare the efficacy of salubrinal with that of our hit compounds, the concentration of compounds required to achieve a 50 % cell survival rate after incubation with thapsigargin (EC50) was determined by measuring cellular ATP levels as a surrogate for cell viability. AMC compounds suppressed thapsigargin-induced cell death more efficiently than salubrinal with lower EC50 levels (Fig. 1e and Table 1). A total of 10 structural analogs of benzyl piperazine benzamide (IA-1 to IA-9 and AMC-05) were tested for effects on CSM14.1 cell viability. Among the newly acquired structural derivative compounds, 1-(4-biphenylylcarbonyl)-4-(2,4,5-trimethoxybenzyl)piperazine (AMC-05) was found to be effective in suppressing thapsigargin-induced cell death with a comparable or better EC50 compared with that of other compounds. Therefore, we have identified a series of small molecules that can more potently protect cells from thapsigargin-induced cell death than salubrinal.
Structure-activity relation analysis
As described above, 14 structural derivative compounds, including AMC compounds, were tested for their efficacy in inhibiting thapsigargin-induced cell death (Table 1). Among these compounds, the best relative survival activity (EC50 = 12.43 ± 1.07 μM) was shown by AMC-01, which contains an ortho-methoxy and a meta-bromo group on its benzyl ring (Fig. 1a and Table 1). By structure-activity relation (SAR) analysis, we found that the introduction of a methoxy group to the benzyl ring of the substituent at the ortho position resulted in increased relative cell survival. Compounds with phenyl substituents on the piperazine benzamide showed promising EC50 values. This relative increase suggests that an ortho methoxy on the benzyl ring and a phenyl moiety on piperazine benzamide play roles in cell survival. To determine whether the ortho methoxy moiety on the benzyl ring was important for this activity, the methoxy group of compound IA-4 was modified. The phenyl moiety on piperazine benzamide was also substituted for a methoxy group (compound IA-2). In compounds with substitutions in the methoxy or phenyl groups, the activity was dramatically reduced. Additionally, compound AMC-05 that contained tri-methoxy groups on the benzyl ring showed relatively less survival activity (EC50 = 35.57 ± 4.28 μM).
AMC compounds protect cells from ER stress-induced cell death
Cell death can be triggered in various ways. To determine the cell death pathway targeted by the hit compounds, we evaluated the protective activities of the compounds in CSM14.1 cells under various cell death-stimulating conditions (Fig. 2). CSM14.1 cells were challenged with tunicamycin (an N-linked glycosylation inhibitor) to compare the effects of an ER stress inducer other than thapsigargin, or with VP16 (a topoisomerase inhibitor that induces DNA damage and mitochondria-mediated cell death), staurosporine (STS, a broad spectrum kinase inhibitor), a combination of tumor necrosis factor alpha (TNF-α) and cyclohexamide (CHX) to induce the receptor-mediated extrinsic cell death pathway, hydrogen peroxide (H2O2) to induce oxidative stress, or paclitaxel (Taxol, a tubulin-stabilizing mitotic inhibitor). The AMC compounds efficiently inhibited tunicamycin-induced cell death (Fig. 2a), but not cell death initiated by VP16, STS, TNF-α/CHX, H2O2, or Taxol (Fig. 2b–f).

Benzyl piperazine benzamide (AMC) compounds selectively inhibit ER stress-induced cell death. CSM14.1 cells were seeded in a 6-well plate for flow cytometry analysis, or in a 96-well plate for assays of ATP content. After 24 h, cells were incubated with DMSO (0.5 %), AMC compounds (25 μM), or control compounds (100 μM salubrinal or 25 μM zVAD). After 2 h of pre-incubation with these above-mentioned compounds, cells were treated with the following cell death-inducing chemical reagents: 10 μg/ml tunicamycin (TUN) for 72 h (a), 50 μM VP16 for 48 h (b), 2.5 μM staurosporine (STS) for 24 h (c), 30 ng/ml TNF-α with 10 μg/ml cyclohexamide (CHX) for 24 h (d), 1 mM hydrogen peroxide (H2O2) for 24 h (e), and paclitaxel 100 nM for 48 h (f). Cellular ATP contents were measured for in VP16, STS, TNF/CHX, H2O2, and Taxol-treated samples; values were normalized to those of DMSO alone-treated cells and are presented as relative survival (percentages of controls). To measure the viability of cells after treatment with tunicamycin, cells were stained with Annexin V-FITC and analyzed by flow cytometry to identify live cells. Assays were performed in duplicate, and data represent survival relative to control cells (DMSO alone) expressed as means ± S.D. (n = 2)
Analysis of unfolded protein response pathways modified by hit compounds
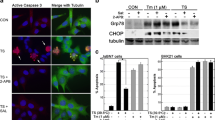
To characterize the cell signaling pathway modified by AMC compounds, we analyzed signaling events associated with the ER stress conditions, by immunoblotting and RT-PCR. Specifically, we analyzed the phosphorylation of eIF2-α at serine 51, c-JUN at serine 73, IRE1 at serine 724, and p38 MAPK at Thr180/Tyr182. We also evaluated overall levels of GRP78, ATF-4 protein expression, cleavage of ATF-6, and XBP-1 messenger RNA (mRNA) transcript splicing. Interestingly, pre-incubation of CSM14.1 cells with AMC-01 (20 μM) increased phosphorylation of eIF2-α. Figure 3a shows that increased phosphorylation of eIF2-α by AMC-01 before artificial ER stress induction (time 0 of TG treatment). Similarly with eIF2-α, p38 MAPK was activated by AMC-01 treatment (Fig. 3a). However, no substantial changes were observed in the cleavage level of ATF-6, phosphorylation of IRE-1, phosphorylation of c-JUN, expression level of GRP 78, splicing of XBP-1 mRNA transcripts or in the levels of ATF-4 proteins when cells were incubated with only this compound (Fig. 3a, b). Bands from immunoblotting data were subjected to densitometry analysis (Fig. 3a, right panel)


Pre-incubation of CSM14.1 cells with AMC-01 induced eIF2-α phosphorylation. a CSM14.1 cells were cultured with DMSO or 25 μM AMC-01 for 2 h, followed by treatment with thapsigargin (10 μM) for the indicated amounts of time. Cell lysates were prepared and analyzed by western blotting using the indicated antibodies. To detect XBP-1 splicing, total RNA samples (1 μg) were analyzed by RT-PCR using a rat XBP-1 primer set. The data is a representative image from three independent experiments. The densitomeric analysis results from western blotting are presented at the right panel. The bar means ± SD (n = 3; * p < 0.05). b CSM14.1 cells were pre-incubated with AMC-01 (25 μM) for 2 h and treated by thapsigargin for the indicated time. After preparing cell lysates, ATF-4 induction was detected by western blotting. c To detect eIF2-α phosphorylation in response to treatment with AMC-01, CSM14.1 cells were incubated with 20 μM AMC-01 for the indicated time. After preparing cell lysates, cell extracts were subjected to western blotting to detect phospho-eIF2-α. Unphosphorylated eIF2-α and α-tubulin were also detected; the latter served as a loading control. d CSM14.1 cells were incubated with AMC compounds or thapsigargin for 2 or 24 h, and cell extracts were subjected to western blotting. Note, eIF2-α phosphorylation induced by AMC compounds was maintained for 24 h. e CSM14.1 cells were pre-incubated with AMC-01 (25 μM) for 2 h and treated with thapsigargin for the indicated time. After preparing cell lysates, eIF2-α kinase activation was examined by western blotting using phosphorylation-specific and normal form antibodies. The data is a representative image from three independent experiments. The densitometric analysis results from western blotting are presented at the right panel. The bar means ± SD (n = 3; *p < 0.05). f CSM14.1 cells were incubated with AMC-01 for 2 or 24 h, and extracts were used for western blotting of PKR and NF-κB p65. The data is a representative image from three independent experiments. The densitometric analysis results from western blotting are presented at the right panel. The bar means ± SD (n = 3; *p < 0.05; **p < 0.01)
The kinetics of the induction of eIF2-α phosphorylation in cells treated with AMC-01 and related compounds were next characterized. As shown in Fig. 3c, phosphorylation of eIF2-α could be induced by AMC-01 in a time-dependent manner. Additionally, we assessed the duration of eIF2-α phosphorylation by AMC compounds in comparison with the ER stress inducer thapsigargin (Fig. 3d). Following the addition of 20 μM compounds to cultures, increased amounts of eIF2-α phosphorylation could be detected within 2 h, which persisted for at least 24 h. By contrast, the phosphorylation of eIF2-α was somewhat more transient in cells treated with thapsigargin, and progressively declined by 24 h (Fig. 3d).
To identify the upstream signaling pathway triggered by AMC compounds, we examined the activation of eIF2-α kinases after incubation with AMC-01. Pre-incubation of cells with AMC-01 did not activate GCN2 or PERK. However, in contrast to GCN2 and PERK, PKR could be activated by AMC-01 pre-incubation, irrespective of the induction of ER stress (Fig. 3e). To confirm that PKR activation could induce pro-survival signaling, we examined the activation of nuclear factor kappa-B (NF-κB) p65, because p65 has been reported to be phosphorylated by activated PKR (Zamanian-Daryoush et al. 2000). As shown in Fig. 3e, NF-κB p65 was activated with phosphorylation by AMC-01 incubation similarly to PKR. Time-dependent activation of both of PKR and p65 were re-examined and presented in Fig. 3f with densitometry analysis.
Correlation of AMC compound-mediated induction of eIF2-α phosphorylation with cytoprotection
The ability of other AMC compounds to induce eIF2-α phosphorylation was also examined by immunoblotting to detect the phosphorylation of eIF2-α. Two structural analog compounds with less cytoprotective characteristics (IA-1 and IA-2) were compared with the AMC compounds. All four cytoprotective AMC compounds induced eIF2-α phosphorylation in a dose-dependent manner, while the inactive analogs IA-1 and IA-2 failed to do so (Fig. 4a). The efficacy of salubrinal was also compared within the same experiment (Fig. 4a, lower). Whereas the AMC compounds induced eIF2-α phosphorylation at concentrations of 10–20 μM, the minimum concentration of salubrinal required for activation was ~30 μM.

AMC compounds induce eIF2-α phosphorylation more potently than salubrinal. a CSM14.1 cells were incubated with the indicated doses of AMC compounds, inactive analog compounds (IA-1 and IA-2), or salubrinal. Cell extracts were prepared and analyzed by western blotting to detect eIF2-α phosphorylation. b Using the same procedures described in (a), cell extracts were prepared and analyzed to detect the ratio of “phospho-eIF2-α/total eIF2-α” using a scanning ELISA. The raw value of phospho-eIF2-α/total eIF2-α from the control extract (DMSO alone) was adjusted to 1, and raw values for other values were normalized to those of the control. Experiments were performed in duplicate with three independent samples. Data represent the means ± SD (n = 2)
To confirm our phospho-protein immunoblotting results, the ability of AMC compounds to stimulate eIF2-α phosphorylation was also examined using enzyme-linked immune sorbent assay (ELISA) (Fig. 4b). All hit compounds induced a dose-dependent increase in the “phospho-eIF2-α/eIF2-α” ratio, whereas the inactive analogs IA-1 and IA-2 failed to significantly increase the phospho-eIF2-α ratio, even for the highest concentration tested (Fig. 4a). Salubrinal also increased levels of phospho-eIF2-α in a dose-dependent manner, but it was less potent compared with the AMC compounds.
CAP-dependent translation can be suppressed by AMC compounds
Inactivation of eIF2-α by serine phosphorylation is known to result in a shutdown of global mRNA translation that can relieve the loading of an incoming peptide into the ER (Donnelly et al. 2013). The eIF2-α protein is a component of the eIF2 complex, which mediates the joining of the methionyl-initiator tRNAMet to the 43S ribosome; the resulting 43S complex can trigger the first step in CAP-dependent mRNA translation (Hinnebusch 2012). Accordingly, we speculated that our chemical probes that could stimulate eIF2-α phosphorylation might also slow the rate of protein synthesis within a cell. To characterize the efficacy of the AMC compounds as eIF2-α-inactivating probes, levels of CAP-dependent translation were examined. CSM14.1 cells were transfected with pRMF vector, which encodes a CAP-dependent Renilla reporter with an IRES-dependent firefly luciferase reporter. We found that CAP-dependent translation was suppressed in CSM14.1 cells with dose-dependent manners by AMC compounds (Fig. 5a).

AMC compounds negatively regulate protein synthesis and CAP-dependent translation. a CSM14.1 cells were transfected with 1 μg pRMF vector for 24 h. After incubation with the indicated dose of AMC compounds, inactive analogs (IA) and salubrinal (Sal.) for 6 h, cell extracts were prepared and subjected to a dual-luciferase assay to evaluate the rate of CAP-dependent translation. Raw values from the control cells were adjusted to 1 and used for normalization of values from compound-treated cells. Experiments were performed in duplicate with three independent samples. Data represent means ± SD (n = 2). b CSM14.1 cells were treated with AMC compounds at the indicated doses, and cell extracts were analyzed for cyclin D depletion by western blotting
To independently verify the reporter gene-based method of assessing CAP-dependent translation, we next evaluated the expression level of Cyclin D1 protein by immunoblotting. Cyclin D1 mRNA transcripts have been characterized as a target for which the translation rate can be affected by eIF2-α activity (Stockwell et al. 2012). When CSM14.1 cells were incubated with AMC compounds, Cyclin D1 protein expression was downregulated in a dose-dependent manner within 4 h of compound exposure. However, the two inactive analog compounds failed to reduce Cyclin D1 expression (Fig. 5b).
Discussion
The synthesis of new proteins requires considerable amounts of cellular energy, and free amino acids are competitively utilized by protein translation and other components of the cell metabolic machinery (Wieser and Krumschnabel 2001). For example, cells with secretory functions, such as primary hepatocytes, are thought to use more than 50 % of their energy for protein synthesis (Pannevis and Houlihan 1992). Accordingly, the machinery that reversibly attenuates protein synthesis is an important cellular adaptation mechanism for maintaining energetic homeostasis, especially during times of cellular stress. The protein translational initiation factor eIF2-α and its upstream kinases represent a physiologically important system for controlling protein translation, and this complex machinery satisfies cellular needs for the careful regulation of protein synthesis (Donnelly et al. 2013). The importance of this cell stress response suggests the need for the development of new chemical probes that can modulate eIF2-α activity, and thereby alter the rate of protein synthesis.
We previously described a cell-based phenotypic screening method to identify small molecule inhibitors of thapsigargin-induced cell death (Kim et al. 2009). Herein, using this approach, we identified structurally related small molecules, including AMC-01, and suggest that they have potential utility as chemical probes that can induce eIF2-α phosphorylation. Our present findings also indicate that AMC-01 and analog compounds can suppress ER stress-induced cell death, which correlates with the induction of eIF2-α phosphorylation at serine residue 51 (Figs. 3 and 4) and the consequential reduced protein synthesis rate and Cyclin D protein depletion (Fig. 5). To investigate the specific molecular role of our novel probe compounds, we used two structurally related control compounds that did not exhibit a cell death-inhibitory function against ER stress-induced cell death (Figs. 1e, 4, and 5). Our findings show that AMC-01 and its analogs are clearly distinguishable from inactive control compounds in the inhibition of ER stress-induced cell death, induction of eIF2-α phosphorylation, and Cyclin D depletion.
Salubrinal, an inhibitor of eIF2-α dephosphorylating phosphatase, has been implicated as an inhibitor of ER stress-induced cell death and was used as a positive control for all of the AMC compound tests that we conducted. We found that AMC-01 had a lower EC50 in cell death inhibition assays (Fig. 1e and Table 1), and a lower range of working concentrations for the induction of eIF2-α phosphorylation compared with salubrinal (Fig. 4). These data suggest that AMC compounds will be more effective chemical probes in future studies of the function of eIF2-α in both stress responses and cell homeostasis.
In our present study, only a fraction of the molecular mechanism whereby AMC compounds can induce the persistent phosphorylation of eIF2-α is addressed. At least four kinases are known to phosphorylate eIF2-α under various cell stress conditions (Donnelly et al. 2013), heme-regulated inhibitor kinase (EIF2AK1/HRI) (Han et al. 2001), RNA-dependent protein kinase (EIF2AK2/PKR) (Galluzzi et al. 2008; Ito et al. 1999; Nakamura et al. 2010; Onuki et al. 2004; Ruvolo et al. 2001), PKR-like ER kinase (EIF2AK3/PERK) (Healy et al. 2009; Szegezdi et al. 2006), and general control non-depressible-2 kinase (EIF2AK4/GCN2) (Deval et al. 2009). Additionally, because we isolated the AMC series of compounds from the ER stress inhibitor screen, we speculate that AMC-01 likely activates one or more signaling pathways related to EIF2A kinases in the unfolded protein response (UPR), which results in eIF2-α phosphorylation. However, our data showed that AMC compounds do not modulate PERK activity, and we also established that ATF-4 was not induced by AMC compounds. These data indicated that the PERK-eIF2-α pathway is not essential for the activities of AMC compounds. Instead, we found that RNA-dependent protein kinase (EIF2AK2/PKR) could be activated by pre-incubation with AMC-01 (Fig. 3). One interesting result is that ATF-4 expression was not upregulated although eIF2-α phosphorylation was enhanced by AMC-01, considering inactivation of eIF2-α by phosphorylation is a precondition for ATF-4 induction (Fig. 3b). However, there are some conditions including UV irradiation which induces eIF2-α phosphorylation without affecting ATF-4 protein expression level (Jiang and Wek 2005), and we can expect the stimuli which AMC compounds give to cell shares similar mechanism of eIF2-α inactivation with these conditions.
Furthermore, we established that NF-κB p65 could be activated by AMC-01, and this finding was comparable to a previous report describing the signaling transduction pathway from PKR to NF-κB (Gil et al. 2000). The activation of GCN2 by AMC compounds was also tested; however, we did not detect marked differences in the activation of GCN2. Conclusively, our data show that AMC compound-induced signaling can proceed via the PKR-eIF2-α pathway rather than the PERK-eIF2-α-ATF4 pathway.
Another possible mechanism whereby AMC compounds result in sustained eIF2-α phosphorylation is the inhibition of eIF2-α dephosphorylation, which occurs by suppressing the activity of a phosphatase, such as protein phosphatase-1; notably, this activity is similar to that of salubrinal (Boyce et al. 2005). Salubrinal has been reported to inhibit PP1-mediated dephosphorylation of eIF2-α, although our data remain insufficient at present to confirm this finding. Future studies that employ a target deconvolution strategy will be needed to address these basic questions regarding the cellular mechanisms activated by the AMC series of compounds.
Although AMC compounds inhibit only ER stress-induced cell death from our test, it is not clear whether these compounds are specific inhibitors against only ER stress-induced cell death. As we defined, the cytoprotective activity of compounds originated from inactivation of eIF2-α, with NF-κB activation by the modulation of PKR activity which is not related with ER stress. And it is possible that the prolonged inactivation of eIF2-α was unlikely to be permanently cytoprotective because the compounds could also suppress protein synthesis. In our preliminary studies, AMC compounds showed no detectable cytotoxicity within 24 h at a concentration of 25 μM. However, the prolonged incubation of cells with AMC-01 could induce death in breast cancer cells (data not shown). We propose that the persistent inactivation of eIF2-α results in the depletion of essential proteins by reducing the rate of protein synthesis. Similarly to various protein synthesis inhibiting compounds, such as cyclohexamide, emetine, and hemoharringtonine, the induction of cell death by prolonged eIF2-α inactivation will represent another interesting area of investigation.
To date, certain findings have been suggested to explain the role of eIF2-α in disease pathogenesis and its control. In cancer cells, the prolonged inactivation of eIF2-α result in reduced viability because of the loss of anti-apoptotic proteins, which are required for the survival of cancer cells (Fritsch et al. 2007). Theoretically, a small molecule that could induce the phosphorylation of eIF2-α could sensitize cancer cells to paclitaxel (Stockwell et al. 2012). In contrast to a previous cancer study (Lee do et al. 2010), activation of PERK, which is a kinase upstream of eIF2-α, was shown to be beneficial in beta-amyloid-induced murine models of neurotoxicity. In future studies, modulation of the activity of eIF2-α represents a potential strategy to study pathogenic conditions that involves EIF2A.
In summary, we have identified a series of eIF2-α-inactivating chemical probes that can more potently sustain eIF2-α phosphorylation than either ER stress-inducing agents or salubrinal. These compounds may provide a useful mechanistic tool for studies of various diseases that involve eIF2-α and its cellular regulators.
References
Balachandran S, Barber GN (2004) Defective translational control facilitates vesicular stomatitis virus oncolysis. Cancer Cell 5:51–65
Boyce M et al (2005) A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307:935–939. doi:10.1126/science.1101902
Chappell SA et al (2000) A mutation in the c-myc-IRES leads to enhanced internal ribosome entry in multiple myeloma: a novel mechanism of oncogene de-regulation. Oncogene 19:4437–4440. doi:10.1038/sj.onc.1203791
Deval C et al (2009) Amino acid limitation regulates the expression of genes involved in several specific biological processes through GCN2-dependent and GCN2-independent pathways. FEBS J 276:707–718. doi:10.1111/j.1742-4658.2008.06818.x
Donnelly N, Gorman AM, Gupta S, Samali A (2013) The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci 70:3493–3511. doi:10.1007/s00018-012-1252-6
Fritsch RM, Schneider G, Saur D, Scheibel M, Schmid RM (2007) Translational repression of MCL-1 couples stress-induced eIF2 alpha phosphorylation to mitochondrial apoptosis initiation. J Biol Chem 282:22551–22562. doi:10.1074/jbc.M702673200
Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G (2008) Viral control of mitochondrial apoptosis. PLoS Pathog 4:e1000018. doi:10.1371/journal.ppat.1000018
Gil J, Alcami J, Esteban M (2000) Activation of NF-kappa B by the dsRNA-dependent protein kinase. PKR involves the I kappa B kinase complex. Oncogene 19:1369–1378. doi:10.1038/sj.onc.1203448
Han AP et al (2001) Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J 20:6909–6918. doi:10.1093/emboj/20.23.6909
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274. doi:10.1038/16729
Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A (2009) Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol 625:234–246. doi:10.1016/j.ejphar.2009.06.064
Hinnebusch AG (2012) Translational homeostasis via eIF4E and 4E-BP1. Mol Cell 46:717–719. doi:10.1016/j.molcel.2012.06.001
Holcik M, Sonenberg N (2005) Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6:318–327. doi:10.1038/nrm1618
Ito T, Yang M, May WS (1999) RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem 274:15427–15432
Jiang HY, Wek RC (2005) GCN2 phosphorylation of eIF2alpha activates NF-kappaB in response to UV irradiation. Biochem J 385:371–380. doi:10.1042/BJ20041164
Kim I et al (2009) Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J Biol Chem 284:1593–1603. doi:10.1074/jbc.M807308200
Koromilas AE (2015) Roles of the translation initiation factor eIF2alpha serine 51 phosphorylation in cancer formation and treatment. Biochim Biophys Acta 1849:871–880. doi:10.1016/j.bbagrm.2014.12.007
Lee do Y et al (2010) Activation of PERK signaling attenuates Abeta-mediated ER stress. PLoS One 5:10.1371/journal.pone.0010489
Ling J, Morley SJ, Traugh JA (2005) Inhibition of cap-dependent translation via phosphorylation of eIF4G by protein kinase Pak2. EMBO J 24:4094–4105. doi:10.1038/sj.emboj.7600868
Nakamura T et al (2010) Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 140:338–348. doi:10.1016/j.cell.2010.01.001
Onuki R et al (2004) An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer's disease. EMBO J 23:959–968. doi:10.1038/sj.emboj.7600049
Pannevis MC, Houlihan DF (1992) The energetic cost of protein synthesis in isolated hepatocytes of rainbow trout (Oncorhynchus mykiss) Journal of comparative physiology B. Biochem, Syst, Environ Physiol 162:393–400
Ruvolo PP, Gao F, Blalock WL, Deng X, May WS (2001) Ceramide regulates protein synthesis by a novel mechanism involving the cellular PKR activator RAX. J Biol Chem 276:11754–11758. doi:10.1074/jbc.M011400200
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789. doi:10.1146/annurev.biochem.73.011303.074134
Silvera D, Formenti SC, Schneider RJ (2010) Translational control in cancer nature reviews. Cancer 10:254–266. doi:10.1038/nrc2824
Sonenberg N, Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. doi:10.1016/j.cell.2009.01.042
Stockwell SR et al (2012) Mechanism-based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PLoS One 7:e28568. doi:10.1371/journal.pone.0028568
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7:880–885. doi:10.1038/sj.embor.7400779
Tabas I, Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13:184–190. doi:10.1038/ncb0311-184
Wang M, Kaufman RJ (2014) The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer 14:581–597. doi:10.1038/nrc3800
Wieser W, Krumschnabel G (2001) Hierarchies of ATP-consuming processes: direct compared with indirect measurements, and comparative aspects. Biochem J 355:389–395
Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR (2000) NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol 20:1278–1290
Acknowledgments
This study was supported by a new scientist research grant from the National Research Foundation of Korea (NRF-2014R1A1A1003831 to KIK), which is funded by the Ministry of Science, ICT, and Future Planning of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Hong, MN., Nam, KY., Kim, K.K. et al. The small molecule ‘1-(4-biphenylylcarbonyl)-4-(5-bromo-2-methoxybenzyl) piperazine oxalate’ and its derivatives regulate global protein synthesis by inactivating eukaryotic translation initiation factor 2-alpha. Cell Stress and Chaperones 21, 485–497 (2016). https://doi.org/10.1007/s12192-016-0677-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-016-0677-5