
Abstract
Myelodysplastic neoplasms (MDS) are defined by cytopenia and morphologic dysplasia originating from clonal hematopoiesis. They are also frequently complicated with diseases caused by immune dysfunction, such as Behçet's disease (BD) and secondary pulmonary alveolar proteinosis (sPAP). MDS with both BD and sPAP is extremely rare, and their prognosis is poor. In addition, haploinsufficiency of the hematopoietic transcription factor gene GATA2 is recognized as a cause of familial MDS and is frequently complicated by sPAP. Herein, we report a case of MDS combined with both BD and sPAP in association with GATA2 deficiency in a Japanese woman. Because she developed progressive leukopenia and macrocytic anemia during BD treatment at the age of 61, she underwent a bone-marrow examination and was diagnosed with MDS. She subsequently developed sPAP. At the age of 63, she underwent allogeneic hematopoietic stem cell transplantation (allo-HSCT). Since allo-HSCT, she has maintained complete remission of MDS as well as the symptoms of BD and sPAP. Furthermore, we performed whole exome sequencing and identified the GATA2 Ala164Thr germline mutation. These findings suggest that patients with MDS, BD and sPAP should be considered for early allo-HSCT.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myelodysplastic neoplasms (MDS) are defined by cytopenia and morphologic dysplasia originating from clonal hematopoiesis (CH) [1]. Mutations associated with CH are often present in circulating granulocytes and monocytes [2, 3]. Therefore, MDS is also frequently complicated by diseases caused by neutrophil and monocyte dysfunction [3, 4]. In particular, MDS with trisomy 8 is occasionally associated with Behçet's disease (BD) [5] and secondary pulmonary alveolar proteinosis (sPAP) [6]. Although allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the only curative therapy available for MDS with sPAP, high transplant-related mortality has been reported [7].
In addition, heterozygous mutations in the hematopoietic transcription factor gene GATA2 have been reported as a cause of familial MDS [8]. GATA2 mutations, regardless of mutation type, appear to cause loss of function of the mutated allele, leading to haploinsufficiency [9]. GATA2 deficiency is involved in immunodeficiency, predisposition to MDS/acute myeloid leukemia (AML), and sPAP [10]. The bone marrow (BM) in patients with GATA2 deficiency is typically hypocellular and contains atypical and micro megakaryocytes [9]. When MDS patients have a familial history of MDS/AML, it should be determined whether they have mutations in GATA2.
Cases of MDS with both BD and sPAP are extremely rare, and its prognosis is known to be poor. In previous reports, two out of three patients died due to severe infection within 6 months of sPAP diagnosis [11]. Herein, we report a case of successful allogeneic HSCT for MDS with BD and sPAP. After allo-HSCT, temporary exacerbation of sPAP was observed but improved with short-term ventilator management. In the present case, MDS, as well as the symptoms of BD and sPAP, remained in remission after allo-HSCT at the time of writing.
Case report
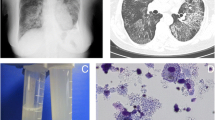
A 58-year-old Japanese woman presented to a local general hospital with chronic diarrhea that had started 1 year previously. She had a history of idiopathic thrombocytopenic purpura during pregnancy, uterine fibroids, stomatitis, and genital ulcers. Her mother had a history of colon cancer, and her daughter had MDS. Colonoscopy revealed multiple ulcers extending from the ileum to the sigmoid colon. Human leukocyte antigen (HLA) analysis was positive for B51 and A26, both of which are associated with BD. She was diagnosed with intestinal BD and administered prednisolone, colchicine, adalimumab, and azathioprine (50 mg/day). However, her abdominal symptoms relapsed after prednisolone was tapered. At the age of 60, she developed uveitis in the right eye. Infliximab (5 mg/kg) was administered every 8 weeks in combination with azathioprine 50 mg/day, which was temporarily effective for abdominal symptoms. Because she developed progressive leukopenia and macrocytic anemia at the age of 61, she was referred to our hospital, where she underwent BM examination. The BM smear showed hypoplastic cellularity with dysplasia of erythroblasts and megakaryocytes (Fig. 1A), and chromosome analysis of BM revealed the presence of trisomy 8 and trisomy 9 as well as X chromosome abnormalities (48, X, i(X)(q10), + 8, + 9 in 12 of the examined 20 cells; 48, idem, dup(1)(q21q32) in 8 of the examined 20 cells). Therefore, the patient was diagnosed with MDS with multilineage dysplasia (MDS–MLD) [Revised international prognostic scoring system (IPSS-R) score, 4.5]. In addition, warts were observed on the extremities (Fig. 1B).

Clinical features of the present case. A Micromegakaryocytes and megaloblastic changes in erythrocytes were observed in the bone marrow smear. B Warts were observed on the extremities. C Colonoscopy revealed multiple ulcers in the ileum and sigmoid colon. The left and middle panels show ulcers in the ileum before transplantation. The right panel shows a scarred ulcer at the ileum end
Moreover, the patient experienced worsening dyspnea 3 months later. Chest CT showed diffuse ground-glass opacities with interlobular septal thickening in both lungs. Bronchoalveolar lavage fluid had a milk-like appearance, and a transbronchial lung biopsy specimen showed that the alveoli were filled with amorphous acellular eosinophilic material. In addition, anti-GM-CSF autoantibody was negative. These findings confirmed the diagnosis of sPAP [12]. Because respiratory failure gradually progressed and could no longer be controlled by whole lung lavage (Fig. 2A), at the age of 63, the patient received allo-HSCT from an HLA-DR1-allele-mismatched unrelated BM donor. The conditioning regimen consisted of fludarabine (180 mg/m2), busulfan (6.4 mg/kg), and total body irradiation (2 Gy). Graft-versus-host disease (GVHD) prophylaxis was initiated with rabbit anti-human thymocyte globulin (2.5 mg/kg), tacrolimus, and short-term methotrexate. BM grafts containing 1.36 × 106/kg CD34 + cells were infused on day 0 (Fig. 2B). Because of persistent mild percutaneous oxygen desaturation, the patient needed low-dose oxygen administration from day 4. Then, respiratory failure progressed rapidly just before neutrophil engraftment (day 22), and she was intubated and managed with mechanical ventilation. Chest CT showed diffuse ground-grass opacities similar to those seen at the initial onset of sPAP (Fig. 2B). However, respiratory failure improved gradually after engraftment, and the patient was extubated on day 30. The absolute neutrophil count recovered to 0.5 × 109/L on day 24, and the platelet count reached 50 × 109/L on day 45.

Chest CT images and clinical course of the present case. A Chest CT images before hematopoietic stem cell transplantation (day 65), at exacerbation of sPAP after transplantation (day 22), and at remission after transplantation (day 87) are shown. B Changes in white blood cell counts, C-reactive protein (CRP), and KL-6 are shown
Furthermore, diarrhea worsened from day 33. Colonoscopy revealed that the ulcer in the ileum, which had been present since the onset of BD, had scarred (Fig. 1C), leading to a suspicion of intestinal GVHD. Cytomegalovirus was not detected in mucosal biopsy specimens from the colon and ileal region. The patient’s diarrhea improved rapidly after methylprednisolone (1 mg/kg/day) was started based on the clinical diagnosis of intestinal GVHD. On day 360 after transplantation, there was no recurrence of MDS, BD, or sPAP (Fig. 2B).
Furthermore, we performed whole exome sequencing (WES) on genomic DNA extracted from the buccal mucosa and BM mononuclear cells collected before allo-HSCT to evaluate gene mutations in MDS with BD and sPAP. As a result, we identified the germline heterozygous mutation of GATA2 (c.490G > A, p.Ala164Thr).
Discussion
Treatment for MDS with BD and sPAP is challenging. To the best of our knowledge, this is the first report of successful allo-HSCT for this disease. We believe that the findings of this case provide new insights into the course and management of the disease.
Hypomethylating agents and whole-lung lavage have limited efficacy for sPAP combined with MDS [6], and the prognosis of MDS complicated by sPAP is worse than that of MDS without sPAP. Therefore, patients with MDS complicated by sPAP should be considered for early allo-HSCT. When pulmonary complications occur in such patients in the early posttransplant period, exacerbation of PAP, infectious pneumonia, and idiopathic pneumonia syndrome are suspected [6, 7]. In the present case, because respiratory failure due to PAP progressed before the period of engraftment, the patient required ventilator management. Fortunately, alveolar lavage was avoided, because the lung field shadows tended to improve after ventilator management was started. As discussed in previous reports, the differentiation of donor-derived hematopoietic stem cells into alveolar macrophages was delayed compared to that into peripheral blood neutrophils [13], and in the present case, alveolar proteinosis may have been exacerbated early in transplantation when MDS clone-derived alveolar macrophages were still present due to preconditioning chemotherapy, radiation, infection, cytokines, and other factors. It is possible that the patient had a late increase in donor-derived alveolar macrophages after engraftment, which may have been alleviated by supportive care. Although whole-lung lavage should not be delayed, whole-lung lavage may not necessarily be performed for exacerbations of PAP after transplantation.
Moreover, the patient did not undergo MDS-specific treatment, such as azacitidine, while awaiting donor selection and transplantation. Azacitidine has an immunomodulatory effect [14] and can potentially induce durable remission in glucocorticoid-refractory autoimmune conditions that are accompanied by MDS, including BD [15]. While it seems reasonable to speculate that azacitidine could also be efficacious in treating sPAP/MDS, there have been no documented cases of sPAP/MDS being successfully managed with azacitidine [6]. We refrained from administering azacitidine to the patient due to the lack of confirmed efficacy and the high risk of infectious adverse events that may impede the feasibility of HSCT.
In the present case, GATA2 germline mutations (Ala164Thr) were detected in buccal mucosa and BM mononuclear cells by WES. GATA2 deficiency is a recently recognized syndrome [10] and presents with a variety of clinical manifestations, such as hematologic disorder, repetitive infection, and pulmonary diseases. There is no association between clinical presentation and type of mutation [10]. A previous report provided a summary of 57 patients with GATA2 deficiency, wherein the prevalent bone-marrow pathology observed was MDS, frequently complicated with trisomy 8 or monosomy 7 [10]. This observation suggests that GATA2 deficiency may trigger genome instability, eventually leading to trisomy 8, although conclusive evidence for a causal relationship is still lacking. The present case had many characteristics consistent with GATA2 deficiency, such as a family history of MDS, warts, and sPAP. The precise function of the GATA2 Ala164Thr protein remains to be elucidated; however, expression from a single allele is clearly insufficient for long-term normal hematopoiesis [16]. Further investigation is required to confirm the functional abnormalities of GATA2 as a result of this mutation.
Furthermore, a standard conditioning regimen in HSCT for GATA2 deficiency has not yet been established. Transplant-related mortality caused by infection and pulmonary complications is a major concern in HSCT for GATA2 deficiency [17, 18]. In the largest prospective case series of 59 adolescent and adult patients with GATA2 mutations undergoing HSCT [19], a busulfan-based regimen coupled with post-transplantation cyclophosphamide (PT/Cy) resulted in the highest GVHD-free overall survival and event-free survival. However, PT/Cy is not available for HSCT using unrelated-BM donors in Japan. In preceding investigations, instances where hypocellularity in the marrow is evident, without overt dysplasia or minimal/borderline dysplasia not meeting WHO criteria for MDS, and without cytogenetic abnormalities, or with trisomy 8 alone, have shown successful engraftment with a regimen of nonmyeloablative or reduced-intensity conditioning (RIC) [19,20,21]. On the other hand, RIC with alemtuzumab in vivo T-cell depletion is acceptable for severe infectious and respiratory complications in patients with GATA2 deficiency [21]. In vivo T-cell depletion provided excellent control of GVHD, even with mismatched donors [21]. Therefore, we selected the RIC regimen and incorporated low-dose TBI and ATG to mitigate the high risk of engraftment failure and acute GVHD due to HLA-DRB1-mismatched transplant. Although TBI is a known risk factor for noninfectious pulmonary complications, we anticipated a rapid recovery of the patient's pulmonary function after engraftment.
In conclusion, MDS patients with BD and sPAP may have mutations in GATA2, and allo-HSCT should be considered before respiratory failure progression due to sPAP. If we can successfully manage transient exacerbations of sPAP after allo-HSCT, long-term survival may be achieved.
Data availability statement
WES analysis data were deposited in the JGA database (accession number JGAS000584).
References
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36:1703–19.
Arends CM, Galan-Sousa J, Hoyer K, Chan W, Jager M, Yoshida K, et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia. 2018;32:1908–19.
Jaiswal S. Clonal hematopoiesis and nonhematologic disorders. Blood. 2020;136:1606–14.
Wolach O, Stone R. Autoimmunity and inflammation in myelodysplastic syndromes. Acta Haematol. 2016;136:108–17.
Wesner N, Drevon L, Guedon A, Fraison JB, Terrier B, Trad S, et al. Gastrointestinal Behcet’s-like disease with myelodysplastic neoplasms with trisomy 8: a French case series and literature review. Leuk Lymphoma. 2019;60:1782–8.
Hashimoto M, Itonaga H, Nannya Y, Taniguchi H, Fukuda Y, Furumoto T, et al. Secondary pulmonary alveolar proteinosis following treatment with azacitidine for myelodysplastic syndrome. Intern Med. 2020;59:1081–6.
Ishii H, Seymour JF, Tazawa R, Inoue Y, Uchida N, Nishida A, et al. Secondary pulmonary alveolar proteinosis complicating myelodysplastic syndrome results in worsening of prognosis: a retrospective cohort study in Japan. BMC Pulm Med. 2014;14:37.
Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–7.
Hsu AP, Johnson KD, Falcone EL, Sanalkumar R, Sanchez L, Hickstein DD, et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood. 2013;121(3830–7):S1-7.
Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809–21.
Handa T, Nakatsue T, Baba M, Takada T, Nakata K, Ishii H. Clinical features of three cases with pulmonary alveolar proteinosis secondary to myelodysplastic syndrome developed during the course of Behcet’s disease. Respir Investig. 2014;52:75–9.
Shimizu H, Sato S, Suzuki T, Sasajima T, Takahata Y, Shinohara N, et al. Intestinal Behcet’s disease complicated by myelodysplastic syndrome and secondary pulmonary alveolar proteinosis: a case report. BMC Gastroenterol. 2021;21:488.
Casado LF, De la Camara R, Granados E, Giron R, Aspa J, Steegmann JL. Reconstitution of alveolar macrophages from donor marrow in allogeneic BMT; a study of variable number tandem repeat regions by PCR analysis of bronchoalveolar lavage specimens. Haematologica. 1999;84:187–9.
Mekinian A, Zhao LP, Chevret S, Desseaux K, Pascal L, Comont T, et al. A phase II prospective trial of azacitidine in steroid-dependent or refractory systemic autoimmune/inflammatory disorders and VEXAS syndrome associated with MDS and CMML. Leukemia. 2022;36:2739–42.
Yilmaz U, Ar MC, Esatoglu SN, Bavunoglu I, Erzin YZ, Hatemi AI, et al. How to treat myelodysplastic syndrome with clinical features resembling Behcet syndrome: a case-based systematic review. Ann Hematol. 2020;99:1193–203.
Rodrigues NP, Janzen V, Forkert R, Dombkowski DM, Boyd AS, Orkin SH, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106:477–84.
Kotmayer L, Romero-Moya D, Marin-Bejar O, Kozyra E, Catala A, Bigas A, et al. GATA2 deficiency and MDS/AML: Experimental strategies for disease modelling and future therapeutic prospects. Br J Haematol. 2022;199:482–95.
Jorgensen SF, Buechner J, Myhre AE, Galteland E, Spetalen S, Kulseth MA, et al. A nationwide study of GATA2 deficiency in Norway-the majority of patients have undergone Allo-HSCT. J Clin Immunol. 2022;42:404–20.
Nichols-Vinueza DX, Parta M, Shah NN, Cuellar-Rodriguez JM, Bauer TR Jr, West RR, et al. Donor source and post-transplantation cyclophosphamide influence outcome in allogeneic stem cell transplantation for GATA2 deficiency. Br J Haematol. 2022;196:169–78.
Grossman J, Cuellar-Rodriguez J, Gea-Banacloche J, Zerbe C, Calvo K, Hughes T, et al. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biol Blood Marrow Transplant. 2014;20:1940–8.
Tholouli E, Sturgess K, Dickinson RE, Gennery A, Cant AJ, Jackson G, et al. In vivo T-depleted reduced-intensity transplantation for GATA2-related immune dysfunction. Blood. 2018;131:1383–7.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (to D.K.), a grant from the Takeda Science Foundation (to D.K.) and The Uehara Memorial Foundation (to T.I.)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Sato, Y., Fukatsu, M., Suzuki, T. et al. Successful allogeneic hematopoietic stem cell transplantation for myelodysplastic neoplasms complicated with secondary pulmonary alveolar proteinosis and Behçet's disease harboring GATA2 mutation. Int J Hematol 118, 642–646 (2023). https://doi.org/10.1007/s12185-023-03603-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-023-03603-0