
Abstract
Chronic lymphocytic leukemia (CLL), the most frequent type of leukemia in adults, is a lymphoproliferative disease characterized by the clonal expansion of mature CD5+ B cells in peripheral blood, bone marrow, and secondary lymphoid tissues. Over the past decade, substantial advances have been made in understanding the pathogenesis of CLL, including the identification of recurrent mutations, and clarification of clonal architectures, transcriptome analyses, and the multistep leukemogenic process. The biology of CLL is now better understood. The present review focuses on recent insights into CLL leukemogenesis, emphasizing the role of genetic lesions, and the multistep process initiating from very immature hematopoietic stem cells. Finally, we also review progress in the study of human B1 B cells, the putative normal counterparts of CLL cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic lymphocytic leukemia (CLL), the most frequent lymphoproliferative disease in adults, is characterized by the clonal expansion of mature CD5+ B cells. CLL accounts for 25–30% of all types of leukemia [1], and typically affects elderly individuals [2]. Next generation sequence (NGS) technologies have clarified the recurrent genetic lesions in CLL and identified the molecular pathways involved in CLL pathogenesis. Studies revealed that CLL genomes exhibit the heterogeneity between patients with CLL and within cells of the same patient [3]. Moreover, CLL leukemogenesis has been described as a multistep process initiating from immature hematopoietic stem cells (HSCs) [4]. In addition, the impressive efficacies of the kinase inhibitor ibrutinib and of the BCL-2 antagonist venetoclax have changed the standard of care for specific subsets of patients with CLL [5]. This review focuses on the recent insights into the CLL leukemogenesis, emphasizing the role of genetic legions, and the various steps involved. In addition, we also introduce the progress in prospective isolation of human B1 cells and their ontogeny. B1 cells are biologically similar to CLL cells, and have been studied as the possible cellular origins of CLL cells.
Biological features and genetic lesions of CLL
CLL is a B cell malignancy, which is characterized by accumulation of clonal mature CD5-expressing B cells in the blood, bone marrow, and lymphoid tissues [6,7,8]. The prevalence of CLL increases dramatically along with age. CLL cells express functional B cell receptors (BCRs) on their cell surfaces [6, 9, 10]. CLL types are divided into two subgroups based on the presence of somatic hypermutations within the variable regions of the immunoglobulin heavy chain (IGHV) genes patients with CLL with mutated IGHV genes (IGHV-M CLL) have a more favorable prognosis than patients with CLL with unmutated IGHV genes (IGHV-UM CLL) [11]. It has been considered that CLL originate from self-reactive B cell precursors, and that the BCR somatic hypermutation status does not indicate the origin of CLL cells [12,13,14].
IGH-related translocations are rare in CLL, but more common in other types of mature B cell malignancies. The most frequent genetic lesions in CLL are deletions of 13q14 (del13q14) (50–60%) [15, 16]. Most del13q14 deletions are monoallelic and more frequently found in the IGHV-M CLL than in IGHV-UM CLL. In general, Del13q14 is associated with a favorable prognosis, but the clinical course of CLL is accelerated in patients with large del13q14 deletions that affect the retinoblastoma gene (RB1) [17]. The acquisition of chromosome 12 (trisomy12) occurs in ~ 15% of patients with CLL [15, 16]. Trisomy 12 was regarded as a genetic lesion for intermediate risk; but a study revealed that the presence of NOTCH1 mutations in patients with trisomy 12 was associated with poor survivals [18]. Moreover, patients with CLL and trisomy 12 have a higher risk for the progression of Richter syndrome (RS) [19,20,21]. The deletions in the 11q22–23 (del11q) chromosomal region are detected in ~ 15% of CLL cases [15, 16, 22], and del11q results in the loss of the ATM gene (tumor suppressor ataxia telangiectasia mutated), which encodes a DNA damage response kinase ATM [23]. About 25% of patients with CLL and del11q deletions harbor mutations in the remaining ATM allele, and the combination of del11q and ATM mutation in CLL is associated with poor prognosis [24]. Deletions in the 17p13 chromosomal locus (del17p) are observed in ~ 10% of the patients [15, 16, 22], and are frequently observed in IGHV-UM CLL [15]. Del17p deletions usually involve the entire short arm of chromosome 17, leading to the loss of the tumor suppressor gene TP53 [25]. Missense mutations in the remaining TP53 allele are found ~ 80% patients with CLL and del17p [26, 27]. Consistent with the inactivation of TP53 genes, patients with del17p exhibit high genomic complexity and poorer overall prognosis than those with wild-type TP53 [15, 25,26,27,28,29].
In addition to the large chromosomal abnormalities described above, advances in the NGS technologies have revealed recurrent driver mutations in CLL such as SF3B1, ATM, TP53, NOTCH1, POT1, CHD2, XPO1, BIRC3, BRAF, MYD88, EGR2, MED12, FBXW7, ASXL1, KRAS, NRAS, MAP2K1,NFKBIE,TRAF3, and DDX3X [16, 30,31,32,33,34].
SF3B1 mutations are the most frequently observed point mutations in CLL (10–15% of cases) [30,31,32]. SF3B1 mutations cause alternative splicing in CLL cells and induce RNA changes affecting multiple CLL-associated pathways [35].
NOTCH1 is also a frequently mutated gene in CLL (~ 10% of cases) [16, 34, 36]. NOTCH1 mutations are preferentially observed in IGHV-UM CLL. Of note, ~ 40% of patients with NOTCH1-mutated CLL harbor a trisomy 12, implying the relevance of these two genetic aberrations in the pathogenesis of CLL [18, 37]. The vast majority of NOTCH1 mutations in CLL increase the nuclear NOTCH intracellular domain through the abrogation of the PEST domain, which are necessary for F-box and WD repeat containing protein7 (FBXW7)-mediated proteasomal degradation of NOTCH1 [3, 34, 38]. Interestingly, FBXW7-inactivating mutations have been found in patients with CLL without NOTCH1 mutations (~ 3% of patients with CLL), indicating an analogous outcome of enhanced NOTCH1 signaling. Moreover, NOTCH1 activation independent of NOTHC1 mutations has been reported in CLL cells [39, 40]. Thus, the activation of the NOTCH1 pathway via multiple mechanisms can be involved in the pathogenesis of CLL [41].
POT1 mutations are found in 3–7% of CLL patients, and frequently observed in IGHV-UM CLL [16, 30, 31, 34, 42]. POT1 plays an important role in the telomere protection [43]. During normal hematopoiesis, POT1 activity is required for maintaining the activity of self-renewing HSCs [44]. POT1 mutations alter the telomeric DNA binding domain, leading to structural aberrations and chromosomal instability [42].
In all, the genetic CLL lesions can be categorized into several biological pathways such as NOTCH1 signaling, BCR signaling, DNA damage response, genome/chromatin structure, RNA and ribosomal processing, inflammatory pathways, NF-κB signaling, cell cycle, and apoptosis [16, 34]. Thus, these deregulated biological pathways coordinately drive CLL leukemogenesis in human (Fig. 1).

Summary of the pathways and molecules involved in the pathogenesis of CLL. These deregulated biological pathways affected by genetic and non-genetic mechanisms coordinately drive the leukemogenesis of CLL. The sizes of the triangle indicate the frequency of mutations reported in CLL
Multistep leukemogenesis of CLL initiating from HSCs
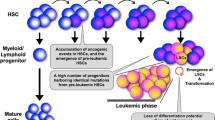
After describing the important molecular pathways involved in the pathogenesis of CLL by NGS studies, we will focus on how such oncogenic events initiate and accumulate during the complex leukemogenesis process. Other types of human leukemia (including acute myeloid leukemia, acute lymphoblastic leukemia, and chronic myeloid leukemia) have HSCs and immature progenitor cells playing roles in their pathogenesis, but CLL has been thought to originate from mature B cells. To trace the origins of human CLL, it is important to note that CLL is not always monoclonal [45, 46]. Moreover, a large cohort study demonstrated that virtually all patients with CLL had prior monoclonal B cell lymphocytosis (MBL) [47]. MBL is a preleukemic state of CLL representing the asymptomatic proliferation of clonal B cells with circulating numbers < 5000/μl [48]. The prevalence of MBL increases with age [47, 49], and it ranges from < 1% [50, 51] to 18% [52]. Of note, human MBL sometimes comprises oligoclonal B cell clones [53,54,55,56,57].
The progression from MBL to CLL reflects a step-wise process, but the stage at which the first oncogenic event occurs remains unknown. The existence of oligoclonal B cell clones in both patients with CLL and MBL suggests that the first oncogenic event may be traced as far back as the progenitor or HSCs. These observations led us to evaluate the primitive HSC fraction in patients with CLL, and we found that the propensity to generate clonal mature B cells is already present in HSCs. CLL cells never directly engrafted in xenograft models; but HSCs derived from patients with CLL gave rise to the abnormal monoclonal or oligoclonal mature B cells in vivo [58]. Moreover, NGS studies confirmed that CD34+ CD19− hematopoietic stem/progenitor cells (HSPCs) and/or myeloid cells from patients with CLL shared identical somatic mutations detected in CLL cells. Such recurrent mutations include NOTCH1, SF3B1, BRAF, TP53, XPO1, MED12, NFKBIE and EGR2 [33, 59, 60]. Whole genome sequence analyses also confirmed shared mutations between MBL/CLL cells and their respective polymorphonuclear cells, suggesting that the acquisition of some somatic mutations occurs before disease onset, likely at the HSCs stage [61]. In addition, the activation of NOTCH1 pathways is deeply involved in CLL leukemogenesis [41], and a study showed that NOTCH1 is aberrantly activated in HSPCs from patients with CLL, regardless of NOTCH1 mutation status (when compared to the HSPC levels in healthy donors), indicating that activation of NOTHC1 is an early event in CLL leukemogenesis that may contribute to the development of aberrant HSPCs in patients with CLL [60]. Consistent with this, advances in the analysis of IGH genes using NGS technology confirmed the presence of independent oligoclonal B cell clones (even in immunophenotypically monoclonal CLL patients) [62]. Thus, the initial oncogenic events occur in human self-renewing HSCs, which then mutate in the multistep leukemogenesis process of CLL. In addition to CLL, studies have clarified that the initial oncogenic events target HSPCs in several human mature lymphoid malignancies [63,64,65,66] as well as in murine models of mature lymphoid malignancies [63, 65, 67,68,69].
These studies have provided the novel steps in the complex leukemogenesis/lymphomagenesis process; cellular stages of tumor-initiation and final transformation are different, and the stage-specific oncogenic events coordinately drive tumor progression. Further studies will help us clarify the molecular mechanisms involved in this step-wise leukemogenesis/lymphomagenesis of the mature lymphoid malignancies.
Understanding B1 cell biology to clarify mechanisms leading to CLL
Next, we will focus on the normal B cell counterparts of CLL. As described above, the initial oncogenic events start and accumulate in HSCs, and such mutated HSCs continuously produce their progeny including mature B cells harboring identical mutations. For the development of MBL, the preleukemic state of CLL, mature B cells derived from such HSCs expand clonally and are maintained while accumulating subsequent oncogenic events that lead to the progression of CLL [4]. The question is which mature B cells are the direct cellular origin of human CLL.
Over the years, different types of B cells have been proposed as the normal counterparts of CLL. BCR signaling can play a critical role in the development of CLL. Of note, it is important to know that CLL cells express a restricted BCR repertoire including antibodies with quasi-identical CDR3 [70,71,72,73]. The striking degree of structural restriction of BCRs in CLL suggests that CLL cells may be driven by recognizing the similar antigens in vivo, and supports the hypothesis that an antigen-driven clonal selection process can be involved in the pathogenesis of CLL [7]. Such antigens may include autoantigens, because BCR of CLL cells exhibit autoreactivity and polyreactivity, suggesting that CLL cells originate from self-reactive B cell precursors [12]. Studies have identified several autoantigens recognized by BCRs of CLL cells [74,75,76]. Moreover, others have demonstrated that BCRs in patients with CLL often have the capacity for autonomous signaling via self-ligation to BCRs independently of ligands [77, 78]. Thus, an important biological feature of CLL is signaling through the autoreactive BCRs. In addition to such autoreactivity of human CLL cells, another important biological character of CLL is the expression of CD5 and IgM.
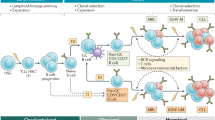
Since these biological features and immunophenotype of CLL cells are very similar to those of mouse B1 cells, B1 cells have been regarded as the possible normal counterparts of CLL cells [79, 80]. B1 cells were first reported as a rare CD5+ B cell subpopulation that secretes IgM [81]. In contrast to conventional B cells (B2 cells), B1 cells were identified at a relatively low frequency in the spleen, but they were abundant in the peritoneal cavity [82, 83]. Importantly, B1 cells differ functionally from B2 cells in their spontaneous secretion of natural IgM that is a more germ-line-like immunoglobulin than that in B2 cells because of their minimal N-region addition, broad reactivity, restricted repertoire, and autoreactivity [84,85,86,87]. Such natural IgM secreted from B1 cells plays an important role in the early defense of bacterial and viral infections [88,89,90]. B1 cells are divided into two subsets according to CD5 expression; CD5+ B1a and CD5− B1b cells [91].
Regarding the ontogeny of B1 cells, B1 cells emerge independently of HSCs during the early embryonic development [82, 92, 93], and they have their own self-renewal capacity [94], whereas B2 cells are derived from HSCs [95]. Studies have shown that mouse B1 cells are also generated from adult HSCs; but the extent to which adult HSCs contribute to B1 cell development, especially to B1a cells, has been debated [95,96,97,98].
Based on these unique biological features of B1 cells, they have been investigated as the cellular origin of CLL development in mouse models. Studies using mouse models have revealed that CLL-like disorders develop efficiently from B1 cells in aged model mice [99,100,101].
Prospective isolation of human B1 cells
Despite the biological similarities between murine B1 cells and CLL cells, human B1 cells have not been intensively investigated as a normal counterpart of CLL due to three reasons: First, about half of the patients with CLL have IGHV-M CLL, and these CLL cells have extensive somatic hypermutations, which is not compatible with the biological character of B1 cells [79, 95]. Second, the normal counterpart of human CLL has been mainly investigated based on gene expression profiling (GEP) analysis [13, 14, 102]. Early GEP studies have revealed a relatively homogeneous GEP irrespective of IGHV mutation status [13, 14], similar to that of human CD27+ memory B cells [13]. A recent GEP analysis by comparing CD5+ CLL cells and prospectively-isolated human B cell subsets suggested that IGHV-M CLL exhibited similar GEP with CD5+CD27+ post-germinal center (GC) subset, whereas GEP of IGHV-UM CLL resembled that of CD5+ CD27− pre-GC B cells [102]. GEP analysis is useful to identify similarities and/or differences among specific cellular populations, but care should be exerted when interpreting the results of GEP analysis. For example, the investigated cellular populations are not always homogeneous. If the analyzed subsets consist of several distinct cellular components, interpretation of results becomes difficult. The third reason is that the characteristics of human B1 cells remain unclear (including their immunophenotype) leading to insufficient information of human B1 GEP. Thus, GEP analyses may not have been adequate to compare CLL cells with human B1 cells.
In 2011, CD20+CD27+CD43+CD70− cells were isolated as human B1 cells, which had murine B1 cell-specific properties such as spontaneous IgM secretion, efficient T cell stimulation, and tonic intracellular signaling [103]. To date, CD19+CD20+CD27+ CD38lo/int CD43+ is regarded as an accurate immunophenotype of human B1 cells [104, 105]. Interestingly, the immunophenotype of CD19+ CD20+ CD27+ human B1 cells is shared with human memory B cells, indicating that human B1 cells were analyzed as part of the human memory B cells in the GEP study showing memory B cells exhibited the most similar GEP with CLL [13]. Therefore, further studies comparing GEP of CLL cells and prospectively-isolated human B1 cells will improve our understanding of the human normal B cell counterparts of CLL.
Human B1 cells are derived from adult HSCs
Since mouse B1 cells emerge independently of HSCs during the early embryonic development, they have their own self-renewal capacity, and they are also generated from adult HSCs [82, 92,93,94,95,96,97,98], a follow-up question is whether human B1 cells can be generated from adult HSCs. Studies to clarify the origin of human B1 cells have been conducted: Xenotransplantation of CD34+ CD38−/lo human HSCs from cord blood and adult BM reconstituted human B1 cells in vivo, and the analysis of the patients who underwent autologous/allogeneic hematopoietic stem cell transplantation (HSCT) showed early development of human B1 cells after HSCT [106]. This study provides evidence that human B1 cells can be generated from self-renewing HSCs; but the possibility of contamination of B1 cells from the transplanted cells cannot be excluded. To overcome the limitations of transplantation analysis, a recent study by Kageyama et al. [107] analyzed B1 cells in paroxysmal nocturnal hemoglobinuria patients harboring somatic PIGA mutations, and found a population of B1 B cells derived from PIGA-mutated adult HSCs. Thus, this unmanipulated analysis of human hematopoiesis provides fresh evidence that a fraction of B1 cells are derived from adult HSCs.
These findings are important when we consider the disease relevance of human B1 cells including the immunodeficiency, auto-immune diseases, and CLL. Somatic mutations accumulate within self-renewing human HSCs and their progeny carry the identical mutations in an age-dependent manner, leading to the emergence of clonal hematopoiesis [108, 109]. The extent to which such mutated HSCs contribute to the production of human B1 cells is unclear, but the fact that human HSCs differentiate into B1 cells in steady state hematopoiesis indicate that age-related clonal hematopoiesis (ARCH) [110] can also involve B1 cells in elderly individuals. Further studies are necessary to assess how ARCH affects human B1 cell function such as spontaneous IgM secretion and efficient T cell stimulation.
Conclusions and perspectives
Advances in NGS technologies have clarified the genetic abnormalities acquired in many types of hematological malignancies. In addition to the identification of recurrent mutations, further understanding of the clonal architectures of hematological malignancies have prompted the search for the origin of these diseases. In this review, we focused on the multistep leukemogenesis of CLL. Given that initial oncogenic events occur and accumulate within HSCs of patients with CLL, the fact that human B1 cells originate from adult HSCs and the common biological features in B1 cells and CLL cells suggests human B1 cells may be the counterparts of CLL cells. The characterization of human B1 cells is still undergoing, and further studies are required to assess the biological significance of human B1 cells especially in the field of immunology and hematology.
References
Ghia P, Ferreri AM, Caligaris-Cappio F. Chronic lymphocytic leukemia. Crit Rev Oncol Hematol. 2007;64(3):234–46.
Dighiero G, Hamblin TJ. Chronic lymphocytic leukaemia. Lancet. 2008;371(9617):1017–29.
Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer. 2016;16(3):145–62.
Kikushige Y, Miyamoto T. Pre-malignant lymphoid cells arise from hematopoietic stem/progenitor cells in chronic lymphocytic leukemia. Int J Hematol. 2015;102(5):528–35.
Hallek M, Shanafelt TD, Eichhorst B. Chronic lymphocytic leukaemia. Lancet. 2018;391(10129):1524–37.
Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–15.
Zenz T, Mertens D, Kuppers R, Dohner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2011;10(1):37–50.
Hallek M, Shanafelt TD, Eichhorst B. Chronic lymphocytic leukaemia. The Lancet. 2018;391(10129):1524–37.
Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: revelations from the B-cell receptor. Blood. 2004;103(12):4389–95.
Caligaris-Cappio F, Ghia P. Novel insights in chronic lymphocytic leukemia: are we getting closer to understanding the pathogenesis of the disease? J Clin Oncol. 2008;26(27):4497–503.
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–54.
Herve M, Xu K, Ng YS, Wardemann H, Albesiano E, Messmer BT, et al. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B cell precursors despite expressing different antibody reactivity. J Clin Invest. 2005;115(6):1636–43.
Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194(11):1625–38.
Rosenwald A, Alizadeh AA, Widhopf G, Simon R, Davis RE, Yu X, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194(11):1639–47.
Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6.
Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525–30.
Ouillette P, Collins R, Shakhan S, Li J, Li C, Shedden K, et al. The prognostic significance of various 13q14 deletions in chronic lymphocytic leukemia. Clin Cancer Res. 2011;17(21):6778–90.
Del Giudice I, Rossi D, Chiaretti S, Marinelli M, Tavolaro S, Gabrielli S, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3):437–41.
Fabbri G, Khiabanian H, Holmes AB, Wang J, Messina M, Mullighan CG, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210(11):2273–88.
Chigrinova E, Rinaldi A, Kwee I, Rossi D, Rancoita PM, Strefford JC, et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood. 2013;122(15):2673–82.
Strati P, Abruzzo LV, Wierda WG, O’Brien S, Ferrajoli A, Keating MJ. Second cancers and Richter transformation are the leading causes of death in patients with trisomy 12 chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(7):420–7.
Wierda WG, O’Brien S, Wang X, Faderl S, Ferrajoli A, Do KA, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol. 2011;29(31):4088–95.
Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197–210.
Skowronska A, Parker A, Ahmed G, Oldreive C, Davis Z, Richards S, et al. Biallelic ATM inactivation significantly reduces survival in patients treated on the United Kingdom Leukemia Research Fund Chronic Lymphocytic Leukemia 4 trial. J Clin Oncol. 2012;30(36):4524–32.
Dohner H, Fischer K, Bentz M, Hansen K, Benner A, Cabot G, et al. p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B-cell leukemias. Blood. 1995;85(6):1580–9.
Zenz T, Krober A, Scherer K, Habe S, Buhler A, Benner A, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112(8):3322–9.
Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E, et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol. 2011;29(16):2223–9.
Yu L, Kim HT, Kasar S, Benien P, Du W, Hoang K, et al. Survival of Del17p CLL Depends on Genomic Complexity and Somatic Mutation. Clin Cancer Res. 2017;23(3):735–45.
Ouillette P, Fossum S, Parkin B, Ding L, Bockenstedt P, Al-Zoubi A, et al. Aggressive chronic lymphocytic leukemia with elevated genomic complexity is associated with multiple gene defects in the response to DNA double-strand breaks. Clin Cancer Res. 2010;16(3):835–47.
Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–506.
Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44(1):47–52.
Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–5.
Damm F, Mylonas E, Cosson A, Yoshida K, Della Valle V, Mouly E, et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014;4(9):1088–101.
Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–24.
Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, et al. Transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell. 2016;30(5):750–63.
Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389–401.
Riches JC, O’Donovan CJ, Kingdon SJ, McClanahan F, Clear AJ, Neuberg DS, et al. Trisomy 12 chronic lymphocytic leukemia cells exhibit upregulation of integrin signaling that is modulated by NOTCH1 mutations. Blood. 2014;123(26):4101–10.
Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–33.
Fabbri G, Holmes AB, Viganotti M, Scuoppo C, Belver L, Herranz D, et al. Common nonmutational NOTCH1 activation in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2017;114(14):E2911–9.
Arruga F, Gizdic B, Serra S, Vaisitti T, Ciardullo C, Coscia M, et al. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia. 2014;28(5):1060–70.
Rosati E, Baldoni S, De Falco F, Del Papa B, Dorillo E, Rompietti C, et al. NOTCH1 aberrations in chronic lymphocytic leukemia. Front Oncol. 2018;8:229.
Ramsay AJ, Quesada V, Foronda M, Conde L, Martinez-Trillos A, Villamor N, et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat Genet. 2013;45(5):526–30.
Lei M, Podell ER, Cech TR. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol. 2004;11(12):1223–9.
Hosokawa K, MacArthur BD, Ikushima YM, Toyama H, Masuhiro Y, Hanazawa S, et al. The telomere binding protein Pot1 maintains haematopoietic stem cell activity with age. Nat Commun. 2017;8(1):804.
Sanchez ML, Almeida J, Gonzalez D, Gonzalez M, Garcia-Marcos MA, Balanzategui A, et al. Incidence and clinicobiologic characteristics of leukemic B-cell chronic lymphoproliferative disorders with more than one B-cell clone. Blood. 2003;102(8):2994–3002.
Kikushige Y, Miyamoto T. Hematopoietic stem cell aging and chronic lymphocytic leukemia pathogenesis. Int J Hematol. 2014;100(4):335–40.
Landgren O, Albitar M, Ma W, Abbasi F, Hayes RB, Ghia P, et al. B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med. 2009;360(7):659–67.
Marti GE, Rawstron AC, Ghia P, Hillmen P, Houlston RS, Kay N, et al. Diagnostic criteria for monoclonal B-cell lymphocytosis. Br J Haematol. 2005;130(3):325–32.
Rawstron AC, Bennett FL, O’Connor SJ, Kwok M, Fenton JA, Plummer M, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med. 2008;359(6):575–83.
Shim YK, Vogt RF, Middleton D, Abbasi F, Slade B, Lee KY, et al. Prevalence and natural history of monoclonal and polyclonal B-cell lymphocytosis in a residential adult population. Cytometry B Clin Cytom. 2007;72(5):344–53.
Rachel JM, Zucker ML, Fox CM, Plapp FV, Menitove JE, Abbasi F, et al. Monoclonal B-cell lymphocytosis in blood donors. Br J Haematol. 2007;139(5):832–6.
Marti GE, Carter P, Abbasi F, Washington GC, Jain N, Zenger VE, et al. B-cell monoclonal lymphocytosis and B-cell abnormalities in the setting of familial B-cell chronic lymphocytic leukemia. Cytometry B Clin Cytom. 2003;52(1):1–12.
Nieto WG, Almeida J, Romero A, Teodosio C, Lopez A, Henriques AF, et al. Increased frequency (12%) of circulating chronic lymphocytic leukemia-like B-cell clones in healthy subjects using a highly sensitive multicolor flow cytometry approach. Blood. 2009;114(1):33–7.
Dagklis A, Fazi C, Sala C, Cantarelli V, Scielzo C, Massacane R, et al. The immunoglobulin gene repertoire of low-count chronic lymphocytic leukemia (CLL)-like monoclonal B lymphocytosis is different from CLL: diagnostic implications for clinical monitoring. Blood. 2009;114(1):26–32.
Lanasa MC, Allgood SD, Volkheimer AD, Gockerman JP, Whitesides JF, Goodman BK, et al. Single-cell analysis reveals oligoclonality among ‘low-count’ monoclonal B-cell lymphocytosis. Leukemia. 2010;24(1):133–40.
Shim YK, Rachel JM, Ghia P, Boren J, Abbasi F, Dagklis A, et al. Monoclonal B-cell lymphocytosis in healthy blood donors: an unexpectedly common finding. Blood. 2014;123(9):1319–26.
Klinger M, Zheng J, Elenitoba-Johnson KS, Perkins SL, Faham M, Bahler DW. Next-generation IgVH sequencing CLL-like monoclonal B-cell lymphocytosis reveals frequent oligoclonality and ongoing hypermutation. Leukemia. 2016;30(5):1055–61.
Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246–59.
Quijada-Alamo M, Hernandez-Sanchez M, Robledo C, Hernandez-Sanchez JM, Benito R, Montano A, et al. Next-generation sequencing and FISH studies reveal the appearance of gene mutations and chromosomal abnormalities in hematopoietic progenitors in chronic lymphocytic leukemia. J Hematol Oncol. 2017;10(1):83.
Di Ianni M, Baldoni S, Del Papa B, Aureli P, Dorillo E, De Falco F, et al. NOTCH1 is aberrantly activated in chronic lymphocytic leukemia hematopoietic stem cells. Front Oncol. 2018;8:105.
Agathangelidis A, Ljungstrom V, Scarfo L, Fazi C, Gounari M, Pandzic T, et al. Highly similar genomic landscapes in monoclonal B-cell lymphocytosis and ultra-stable chronic lymphocytic leukemia with low frequency of driver mutations. Haematologica. 2018;103(5):865–73.
Brazdilova K, Plevova K, Skuhrova Francova H, Kockova H, Borsky M, Bikos V, et al. Multiple productive IGH rearrangements denote oligoclonality even in immunophenotypically monoclonal CLL. Leukemia. 2018;32(1):234–6.
Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1):25–38.
Chung SS, Kim E, Park JH, Chung YR, Lito P, Teruya-Feldstein J, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med. 2014;6(238):238ra71.
Horton SJ, Giotopoulos G, Yun H, Vohra S, Sheppard O, Bashford-Rogers R, et al. Early loss of Crebbp confers malignant stem cell properties on lymphoid progenitors. Nat Cell Biol. 2017;19(9):1093–104.
Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46(2):171–5.
Green MR, Vicente-Duenas C, Romero-Camarero I, Long Liu C, Dai B, Gonzalez-Herrero I, et al. Transient expression of Bcl6 is sufficient for oncogenic function and induction of mature B-cell lymphoma. Nat Commun. 2014;02(5):3904.
Vicente-Duenas C, Fontan L, Gonzalez-Herrero I, Romero-Camarero I, Segura V, Aznar MA, et al. Expression of MALT1 oncogene in hematopoietic stem/progenitor cells recapitulates the pathogenesis of human lymphoma in mice. Proc Natl Acad Sci USA. 2012;109(26):10534–9.
Vicente-Duenas C, Romero-Camarero I, Gonzalez-Herrero I, Alonso-Escudero E, Abollo-Jimenez F, Jiang X, et al. A novel molecular mechanism involved in multiple myeloma development revealed by targeting MafB to haematopoietic progenitors. EMBO J. 2012;31(18):3704–17.
Ghiotto F, Fais F, Valetto A, Albesiano E, Hashimoto S, Dono M, et al. Remarkably similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J Clin Invest. 2004;113(7):1008–16.
Messmer BT, Albesiano E, Efremov DG, Ghiotto F, Allen SL, Kolitz J, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med. 2004;200(4):519–25.
Tobin G, Thunberg U, Karlsson K, Murray F, Laurell A, Willander K, et al. Subsets with restricted immunoglobulin gene rearrangement features indicate a role for antigen selection in the development of chronic lymphocytic leukemia. Blood. 2004;104(9):2879–85.
Widhopf GF 2nd, Rassenti LZ, Toy TL, Gribben JG, Wierda WG, Kipps TJ. Chronic lymphocytic leukemia B cells of more than 1% of patients express virtually identical immunoglobulins. Blood. 2004;104(8):2499–504.
Chu CC, Catera R, Hatzi K, Yan XJ, Zhang L, Wang XB, et al. Chronic lymphocytic leukemia antibodies with a common stereotypic rearrangement recognize nonmuscle myosin heavy chain IIA. Blood. 2008;112(13):5122–9.
Hoogeboom R, van Kessel KP, Hochstenbach F, Wormhoudt TA, Reinten RJ, Wagner K, et al. A mutated B cell chronic lymphocytic leukemia subset that recognizes and responds to fungi. J Exp Med. 2013;210(1):59–70.
Hoogeboom R, Wormhoudt TA, Schipperus MR, Langerak AW, Dunn-Walters DK, Guikema JE, et al. A novel chronic lymphocytic leukemia subset expressing mutated IGHV3-7-encoded rheumatoid factor B-cell receptors that are functionally proficient. Leukemia. 2013;27(3):738–40.
Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309–12.
Binder M, Muller F, Frick M, Wehr C, Simon F, Leistler B, et al. CLL B-cell receptors can recognize themselves: alternative epitopes and structural clues for autostimulatory mechanisms in CLL. Blood. 2013;121(1):239–41.
Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117(6):1781–91.
Darwiche W, Gubler B, Marolleau JP, Ghamlouch H. Chronic lymphocytic leukemia B-cell normal cellular counterpart: clues from a functional perspective. Front Immunol. 2018;9:683.
Hayakawa K, Hardy RR, Parks DR, Herzenberg LA. The “Ly-1 B” cell subpopulation in normal immunodefective, and autoimmune mice. J Exp Med. 1983;157(1):202–18.
Hayakawa K, Hardy RR, Herzenberg LA, Herzenberg LA. Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J Exp Med. 1985;161(6):1554–68.
Hardy RR, Hayakawa K, Parks DR, Herzenberg LA, Herzenberg LA. Murine B cell differentiation lineages. J Exp Med. 1984;159(4):1169–88.
Gu H, Forster I, Rajewsky K. Sequence homologies, N sequence insertion and JH gene utilization in VHDJH joining: implications for the joining mechanism and the ontogenetic timing of Ly1 B cell and B-CLL progenitor generation. EMBO J. 1990;9(7):2133–40.
Hardy RR, Carmack CE, Shinton SA, Riblet RJ, Hayakawa K. A single VH gene is utilized predominantly in anti-BrMRBC hybridomas derived from purified Ly-1 B cells: definition of the VH11 family. J Immunol. 1989;142(10):3643–51.
Pennell CA, Mercolino TJ, Grdina TA, Arnold LW, Haughton G, Clarke SH. Biased immunoglobulin variable region gene expression by Ly-1 B cells due to clonal selection. Eur J Immunol. 1989;19(7):1289–95.
Forster I, Gu H, Rajewsky K. Germline antibody V regions as determinants of clonal persistence and malignant growth in the B cell compartment. EMBO J. 1988;7(12):3693–703.
Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J Exp Med. 2000;192(2):271–80.
Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J Exp Med. 1998;188(12):2381–6.
Haas KM, Poe JC, Steeber DA, Tedder TF. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23(1):7–18.
Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. 2011;11(1):34–46.
Kobayashi M, Shelley WC, Seo W, Vemula S, Lin Y, Liu Y, et al. Functional B-1 progenitor cells are present in the hematopoietic stem cell-deficient embryo and depend on Cbfbeta for their development. Proc Natl Acad Sci USA. 2014;111(33):12151–6.
Yoshimoto M, Montecino-Rodriguez E, Ferkowicz MJ, Porayette P, Shelley WC, Conway SJ, et al. Embryonic day 9 yolk sac and intra-embryonic hemogenic endothelium independently generate a B-1 and marginal zone progenitor lacking B-2 potential. Proc Natl Acad Sci USA. 2011;108(4):1468–73.
Herzenberg LA. B-1 cells: the lineage question revisited. Immunol Rev. 2000;175:9–22.
Montecino-Rodriguez E, Dorshkind K. B-1 B cell development in the fetus and adult. Immunity. 2012;36(1):13–21.
Duber S, Hafner M, Krey M, Lienenklaus S, Roy B, Hobeika E, et al. Induction of B-cell development in adult mice reveals the ability of bone marrow to produce B-1a cells. Blood. 2009;114(24):4960–7.
Ghosn EE, Yamamoto R, Hamanaka S, Yang Y, Herzenberg LA, Nakauchi H, et al. Distinct B-cell lineage commitment distinguishes adult bone marrow hematopoietic stem cells. Proc Natl Acad Sci USA. 2012;109(14):5394–8.
Huang CA, Henry C, Iacomini J, Imanishi-Kari T, Wortis HH. Adult bone marrow contains precursors for CD5 + B cells. Eur J Immunol. 1996;26(10):2537–40.
Hayakawa K, Formica AM, Brill-Dashoff J, Shinton SA, Ichikawa D, Zhou Y, et al. Early generated B1 B cells with restricted BCRs become chronic lymphocytic leukemia with continued c-Myc and low Bmf expression. J Exp Med. 2016;213(13):3007–24.
Niss Arfelt K, Barington L, Benned-Jensen T, Kubale V, Kovalchuk AL, Daugvilaite V, et al. EBI2 overexpression in mice leads to B1 B-cell expansion and chronic lymphocytic leukemia-like B-cell malignancies. Blood. 2017;129(7):866–78.
Hayakawa K, Formica AM, Colombo MJ, Shinton SA, Brill-Dashoff J, Morse Iii HC, et al. Loss of a chromosomal region with synteny to human 13q14 occurs in mouse chronic lymphocytic leukemia that originates from early-generated B-1 B cells. Leukemia. 2016;30(7):1510–9.
Seifert M, Sellmann L, Bloehdorn J, Wein F, Stilgenbauer S, Durig J, et al. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med. 2012;209(12):2183–98.
Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20 + CD27 + CD43 + CD70. J Exp Med. 2011;208(1):67–80.
Quach TD, Rodriguez-Zhurbenko N, Hopkins TJ, Guo X, Hernandez AM, Li W, et al. Distinctions among circulating antibody-secreting cell populations, including B-1 cells. Human Adult Peripheral Blood J Immunol. 2016;196(3):1060–9.
Rodriguez-Zhurbenko N, Quach TD, Hopkins TJ, Rothstein TL, Hernandez AM. Human B-1 cells and B-1 cell antibodies change with advancing age. Front Immunol. 2019;10:483.
Quach TD, Hopkins TJ, Holodick NE, Vuyyuru R, Manser T, Bayer RL, et al. Human B-1 and B-2 B cells develop from Lin-CD34 + CD38lo stem cells. J Immunol. 2016;197(10):3950–8.
Kageyama Y, Miwa H, Tawara I, Ohishi K, Masuya M, Katayama N. A population of CD20(+)CD27(+)CD43(+)CD38(lo/int) B1 cells in PNH are missing GPI-anchored proteins and harbor PIGA mutations. Blood. 2019;134(1):89–92.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–98.
Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–87.
Shlush LI. Age-related clonal hematopoiesis. Blood. 2018;131(5):496–504.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Kikushige, Y. Pathophysiology of chronic lymphocytic leukemia and human B1 cell development. Int J Hematol 111, 634–641 (2020). https://doi.org/10.1007/s12185-019-02788-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-019-02788-7