
Abstract
We report the final results from a multicenter, open-label phase I study of carfilzomib plus lenalidomide and dexamethasone in Japanese patients with heavily pretreated relapsed and/or refractory multiple myeloma (RRMM). Twenty-six RRMM patients were enrolled and received a median of 4.0 prior regimens; 12/26 patients (46.2%) completed the planned 18 administration cycles (mean number of cycles: 14.5 ± 4.9). The safety profile was consistent with that of previous carfilzomib studies. All patients experienced adverse events (AEs), but no new safety concerns were observed. The most common grade ≥ 3 AEs (incidence: ≥ 10%) were lymphocyte count decreased (46.2%), platelet count decreased (42.3%), and neutrophil count decreased (34.6%). The overall response rate was 88.5% (23/26; 90% confidence interval: 72.8–96.8). Complete response (CR) or better was achieved by 30.8% of patients compared with 3.8% in the interim analysis. The median time to CR or better response was 9.4 months. Median progression-free survival and duration of response were 19.5 months and 20.3 months, respectively. Median overall survival was not reached. Long-term administration of carfilzomib produced deep response and long-term disease control. The combination of carfilzomib plus lenalidomide and dexamethasone was well tolerated and showed promising clinical efficacy for heavily pretreated RRMM patients.
Clinical trial registration
This clinical trial was registered in the database clinicaltrials.jp (clinical trial registration number: Japic CTI 142677).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recently, various novel therapies, such as proteasome inhibitors and immunomodulators, were approved for the treatment of multiple myeloma (MM) [1, 2]. As a result of the introduction of such new drugs, the life expectancy of patients with MM has increased. Improvement of overall survival (OS) of newly diagnosed patients has been seen in the last decade [3]. Despite the efficacy of these and other novel treatments, MM remains incurable for many patients who experience relapse, even after a period of disease control or remission [1, 3, 4]. Thus, there is an ongoing need for the development of novel therapies to be used as single agents or in combination with existing agents. Moreover, additional research is required to optimize treatment sequences and combinations.
Carfilzomib is a next-generation, epoxyketone proteasome inhibitor [5, 6] that binds selectively and irreversibly to the 20S proteasome, resulting in the accumulation of proteasome substrates and leading to growth arrest and tumor cell apoptosis [6]. Several clinical trials have reported a favorable safety and efficacy profile for carfilzomib monotherapy, the two-drug combination of carfilzomib and dexamethasone, and the three-drug combination of carfilzomib, lenalidomide, and dexamethasone (CLd). A phase III study (ASPIRE) evaluated CLd and showed that this combination resulted in a 31% decrease in the risk of disease progression or death compared with lenalidomide and dexamethasone, and an increase of 8.7 months in the median progression-free survival (PFS) of patients with relapsed MM [7]. In a phase III, head-to-head comparison study with bortezomib (ENDEAVOR), carfilzomib provided a significant and clinically meaningful reduction in the risk of progression compared with bortezomib and dexamethasone [8]. Based on this evidence, carfilzomib was approved by regulatory agencies for the treatment of relapsed and/or refractory MM (RRMM) throughout the world. Moreover, recent results of the ASPIRE [9] and the ENDEAVOR [10] studies expressed significant and clinically meaningful OS improvement vs standard therapies.
In Japan, carfilzomib administered as monotherapy, in combination with dexamethasone (two-drug combination), and also in combination with lenalidomide plus dexamethasone (three-drug combination) has been studied since 2010 and shows promising results for Japanese RRMM patients [11,12,13,14].
The objective of the present study was to assess the safety, tolerability, and efficacy of CLd in heavily pretreated Japanese patients with RRMM. The interim results of this study have been published at the short-term follow-up with a mean of 4.5 cycles administered [13]. This manuscript reports the final results of the long-term follow-up with a mean of 14.5 cycles administered.
Materials and methods
Study design and setting
Details of the study design have been published in the interim analyses report [13]. Briefly, this multicenter, open-label phase I study was conducted in nine centers in Japan. Patients were enrolled between November 2014 and March 2015, and follow-up was completed on February 20, 2017. This trial was registered at clinicaltrials.jp: Japic CTI 142677. Ethical approval was granted by the institutional review boards at each participating site, and all patients provided written informed consent. The study was conducted in accordance with the principles of the Declaration of Helsinki.
Patients
Details of the inclusion and exclusion criteria have also been reported in the interim analyses report [13]. Briefly, the study enrolled male and female patients diagnosed with symptomatic MM, aged ≥ 20 years with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0–2, who had previously received at least one treatment and had relapsed during the last treatment. Patients previously treated with lenalidomide and dexamethasone were eligible if they demonstrated tolerability to the therapy.
Interventions
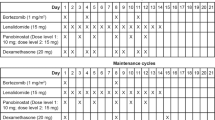
The carfilzomib dosage and administration were based on the ASPIRE study [7, 9]. Treatment lasted a maximum of 18 cycles each consisting of 28 days. During cycles 1–12, carfilzomib was administered as a 10 min intravenous infusion on days 1, 2, 8, 9, 15, and 16. In cycle 1, carfilzomib was administered at a dose of 20 mg/m2 on days 1 and 2 and at a dose of 27 mg/m2 on days 8, 9, 15, and 16 of cycle 1 and thereafter. During cycles 13–18, carfilzomib was administered on days 1, 2, 15, and 16. Lenalidomide was administered orally at a dose of 25 mg on days 1–21 of each 28-day cycle. Dexamethasone was administered orally at a dose of 40 mg on days 1, 8, 15, and 22 of each 28-day cycle. If the dexamethasone dosing day coincided with the carfilzomib dosing day, dexamethasone was administered 30 min to 4 h before carfilzomib administration. Pre- and post-treatment hydration with 250–500 mL of physiological saline (or other appropriate infusion product) were given during cycle 1. Patients received antiviral and antibacterial prophylaxis during cycle 1, and antithrombotic prophylaxis during exposure to lenalidomide.
Endpoints
Tolerability was evaluated in the first six patients based on the evaluation of the transition rate to the extension period and on all adverse events (AEs) (graded using the Common Terminology Criteria for Adverse Events version 4.0 defined by the Japan Clinical Oncology Group [CTCAE v4.0-JCOG]) occurring during cycle 1, including hematologic and non-hematologic toxicities, for which a relationship with the study drug could not be ruled out. Details of the criteria for tolerability and transition to the extension period have been described previously [13].
Safety outcomes included AEs, adverse drug reactions (drug-related AEs), laboratory values, vital signs and 12-lead electrocardiography, transition rate to the extension period, and AEs that met the tolerability evaluation criteria.
Efficacy endpoints included overall response rate (ORR), OS, PFS, time to progression, duration of response (DOR), and clinical benefit rate. Treatment responses and disease progression were assessed by investigators based on the central laboratory results. The efficacy and safety evaluation committee reviewed the investigator assessments.
Chromosome and monoclonal protein analysis
Bone marrow samples for chromosomal analysis were collected during the screening period. Chromosome analysis was performed using fluorescence in situ hybridization to detect t(4;14), t(14;16), and t(11;14) translocations and deletion of chromosome 17, and G-banding to detect hypodiploid of screened plasma cells.
Statistical methods
Details of the sample size calculations have been published previously [13]. Briefly, the planned sample size was 26 subjects, assuming an expected response rate of 87.1% in the CLd cohort in the ASPIRE study [7]. The threshold response rate was 66.7% with at least 70% power on a one-sided exact test and a level of significance of 5.0%, while accounting for a drop-out rate of 4%. A minimum sample size of six subjects was planned for the tolerability evaluation according to the Guidelines for Clinical Evaluation Methods of Antimalignant Tumor Drugs. Safety analyses were conducted in all patients who received at least one dose of study treatment. For efficacy measures, the ORR and its 90% confidence interval (CI) (Clopper–Pearson method) were calculated, and the median PFS and OS were estimated by the Kaplan–Meier method. All statistical analyses were conducted using SAS version 9.3 and 9.4 (SAS Institute, Cary, NC, USA).
Results
Patients
The detailed baseline characteristics have been described in the interim analyses report [13]. Briefly, a total of 26 patients were enrolled, and all were included in the safety and efficacy analysis. Fifty percent of patients were male, and patients had a median age of 64.0 (range, 38–81) years. Overall, 61.5% had an ECOG PS of 0, 76.9% had Revised International Staging System stage 2, 53.8% had received ≥ 4 prior regimens with a median of 4.0 (range, 1–10) regimens, 88.5% had received prior bortezomib therapy, and 61.5% had received prior lenalidomide therapy. High-risk cytogenetics were detected in 53.8% of patients and were defined as positive for del(17p) in ≥ 20% of screened plasma cells, t(4;14), t(14;16), or hypodiploidy.
A total of 12/26 patients (46.2%) completed the planned 18 cycles of carfilzomib administration. In the interim analysis, the mean number of cycles administered was 4.5 ± 1.6 cycles, and it was 14.5 ± 4.9 cycles in a total of 26 patients in this final analysis. The median duration of exposure to each study drug (carfilzomib, lenalidomide, and dexamethasone) was 16.7 months (range, 0.3–21.9), 16.9 months (range, 0.7–22.1), and 17.0 months (range, 0.3–22.1), respectively, and the median relative dose intensities were 92.5% (range, 47.1–100.0), 80.8% (range, 27.4–100.0), and 96.5% (range, 35.2–100.0), respectively. The median duration of follow-up was 18.9 months.
Safety and tolerability
One patient out of the first six patients enrolled experienced an AE which met the criteria for tolerability evaluation. The event was grade 3 upper respiratory tract infection. All of these six patients proceeded to cycle 2. The tolerability of CLd combination for Japanese RRMM patients was confirmed.
All patients (26 of 26) experienced any grade AE, and 84.6% (22 of 26) of patients experienced a grade ≥ 3 AE. Table 1 shows all grade AEs and grade ≥ 3 AEs. The most common AEs of any grade with an incidence of ≥ 20% at the final analysis were lymphocyte count decreased (57.7%), platelet count decreased (57.7%), neutrophil count decreased (46.2%), nasopharyngitis (42.3%), white blood cell count decreased (42.3%), hyperglycemia (42.3%), and hypophosphatemia (42.3%). At the final analysis, the most common grade ≥ 3 AEs with an incidence of ≥ 10% were lymphocyte count decreased (46.2%), platelet count decreased (42.3%), and neutrophil count decreased (34.6%). The incidence of grade ≥ 3 neutrophil count decreased was increased when comparing the interim and final values (final vs interim: 34.6% vs 11.5%).
Six patients (23.1%) experienced a serious AE. The events were sepsis, pneumonia, pneumonia influenza, respiratory tract infection, necrotizing fasciitis, pneumonia aspiration, upper respiratory tract inflammation, dehydration, and back pain in one subject each (3.8%). One patient experienced necrotizing fasciitis, sepsis, and back pain, and another patient experienced both pneumonia and upper respiratory tract inflammation.
Two patients (7.7%) died during the study treatment period or within 30 days after the last dose. One was a 72-year-old male patient who died from sepsis on day 387; the other was an 81-year-old female patient who died of aspiration pneumonia on day 123. The causal relationship between sepsis and the study drug could not be ruled out.
Efficacy
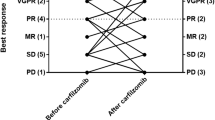
The ORR was 88.5% (23 of 26) (90% CI: 72.8–96.8). The percentage of patients who achieved complete response (CR) or better response was 30.8% (eight of 26), and 46.2% (12 of 26) of patients achieved very good partial response (VGPR) or better response. The clinical benefit rate was 96.2% (25 of 26) (90% CI: 83.0–99.8). Treatment responses are summarized in Table 2. Patients experienced a median PFS of 19.5 months (90% CI: 13.0–22.3) (Fig. 1) and a median time to progression of 19.5 months (90% CI: 14.0–22.3). Median OS was not reached in this study (Fig. 2). The time to CR or better response is shown in Fig. 3 and was 9.4 months (90% CI: 5.3–14.7) among eight complete responders (defined as CR and stringent CR). Median DOR was 20.3 months (90% CI: 12.7–20.3).

Kaplan–Meier curve showing progression-free survival. CI confidence interval, PFS progression-free survival

Kaplan–Meier curve showing overall survival. CI confidence interval, OS overall survival, NE not evaluable

Time to CR or better response. CR complete response, CI confidence interval
A spider plot of changes in M protein level during treatment is shown in Fig. 4. One patient was excluded from this analysis because of the limited number of time points where M protein was measurable.

Spider plot of changes in M protein level during the treatment. The dashed line shows the change in urine M protein in Bence Jones protein-type subjects. Gray squares indicate time points at which changes in the M protein level exceeded 100%
In the subgroup analysis for patients with high-risk cytogenetics, a VGPR or better response was observed in 35.7% (90% CI: 15.3–61.0) of patients in the high-risk subgroup (n = 14) and 58.3% (90% CI: 31.5–81.9) of patients in the standard-risk subgroup (n = 12). The median PFS was not reached (90% CI: 13.6–not evaluable) in the high-risk subgroup, whereas it was 19.5 months (90% CI: 9.0–22.3) in the standard-risk subgroup.
Discussion
This is the first study to evaluate the safety, tolerability, and efficacy of the combination of CLd in heavily pretreated Japanese RRMM patients. Our findings of an ORR of 88.5% and median PFS of 19.5 months, with a favorable safety profile are promising, considering patients enrolled in this study were heavily pretreated and had received a median of four previous treatment regimens. As shown in the spider plot, M protein decreased in a prompt and continuous manner during the treatment period. Even patients who had high-risk cytogenetics could have achieved a VGPR or better response.
Compared with the interim analysis, the proportion of patients who achieved CR or better increased from 3.8 to 30.8%, and the proportion of patients who achieved VGPR or better increased from 23.1 to 46.2% in this final analysis. The mean treatment duration at the final analysis was 14.5 cycles, compared with 4.5 cycles at the interim analysis [13]. These findings suggest that long-term treatment with carfilzomib can induce a deep response. The median time to CR or better response was 9.4 months. These data suggest that continuing carfilzomib treatment for approximately ten cycles is important to maximize the efficacy of this combination regimen. Regarding the safety profile, although nasopharyngitis (42.3% vs 11.5%), neutrophil count decreased (46.2% vs 26.9%), white blood cell count decreased (42.3% vs 30.8%), and upper respiratory tract inflammation (23.1% vs 11.5%) were increased by more than 10% compared with the interim analysis, these events were manageable. Additionally, the fact that 12 out of 26 patients completed the planned 18 cycles of treatment supports the favorable safety profile of this combination regimen. Moreover, the safety profile was consistent with that reported in previous studies with carfilzomib, and no new safety concerns were observed. We have concluded that the safety profile of this combination regimen is favorable and suitable for long-term administration.
The efficacy profile observed in this study was consistent with that of the ASPIRE study [7, 15, 16]. The ORR in the current study was similar to that in the ASPIRE study (88.5% vs 87.1%, respectively). Fewer patients had VGPR or better in the present study compared with the ASPIRE study (46.2% vs 69.9%, respectively). The PFS was longer in the ASPIRE study than in the current study (26.3 months vs 19.5 months, respectively). These differences may be influenced by the patient population (median 2.0 prior regimens in the ASPIRE study vs 4.0 in the present study), the follow-up duration (median 32.3 months in the ASPIRE study vs 18.9 months in the present study), and the difference in the treatment duration with lenalidomide and dexamethasone. In the present study, administration of lenalidomide and dexamethasone was limited to 18 cycles. Conversely, in the ASPIRE study, these drugs were continued until progressive disease occurred. Moreover, the safety profile of this study was also consistent with that reported in the ASPIRE study, and no new safety concerns were observed.
Recently, several phase III trials evaluating three-drug combinations were reported. Ixazomib plus lenalidomide and dexamethasone resulted in a median PFS of 20.6 months [17]. Elotuzumab plus lenalidomide and dexamethasone resulted in a median PFS of 19.4 months [18]. Daratumumab plus lenalidomide and dexamethasone did not achieve the median PFS at the interim analysis; however, the PFS at 1 year among those patients was 83.2% [19]. Regarding Japanese patient data of three-drug combinations, 48% of patients treated with elotuzumab plus lenalidomide and dexamethasone achieved PFS at 2 years [20], and the combination of bortezomib, lenalidomide, and dexamethasone resulted in a median PFS of 9.5 months [21]. Although there are several limitations when comparing clinical data from different trials, the efficacy data of the present study can be considered comparable with previously reported data.
Limitations
This study has several limitations, particularly the small number of patients enrolled, the lack of a comparator, and the inclusion of Japanese patients only, which limits the generalizability of the results. However, this study provides critical data on ORR, PFS, and safety that can be compared with other trials. Furthermore, this study provides valuable data on patients with high-risk cytogenetics.
Conclusions
We conclude that the combination of CLd was well tolerated and resulted in promising clinical efficacy for heavily pretreated RRMM patients. Long-term treatment could maximize the clinical response of this combination, and it is important to continue the treatment as long as possible.
Recently, CLd at a dose of 20/36 mg/m2 showed promising results for newly diagnosed MM [22,23,24]. Future research should focus on evaluating the efficacy and safety of CLd at a higher dose (20/36 mg/m2) in treatment-naïve patients.
References
Dingli D, Ailawadhi S, Bergsagel PL, Buadi FK, Dispenzieri A, Fonseca R, et al. Therapy for relapsed multiple myeloma: guidelines from the Mayo stratification for myeloma and risk-adapted therapy. Mayo Clin Proc. 2017;92:578–98.
Gonsalves WI, Milani P, Derudas D, Buadi FK. The next generation of novel therapies for the management of relapsed multiple myeloma. Future Oncol. 2017;13:63–75.
Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–20.
Mohty B, El-Cheikh J, Yakoub-Agha I, Avet-Loiseau H, Moreau P, Mohty M. Treatment strategies in relapsed and refractory multiple myeloma: a focus on drug sequencing and ‘retreatment’ approaches in the era of novel agents. Leukemia. 2012;26:73–85.
Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120:2817–25.
Steiner RE, Manasanch EE. Carfilzomib boosted combination therapy for relapsed multiple myeloma. Onco Targets Ther. 2017;10:895–907.
Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142–52.
Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hájek R, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17:27–38.
Siegel DS, Dimopoulos MA, Ludwig H, Facon T, Goldschmidt H, Jakubowiak A, et al. Improvement in overall survival with carfilzomib, lenalidomide, and dexamethasone in patients with relapsed or refractory multiple myeloma. J Clin Oncol. 2018;36:728–34.
Dimopoulos MA, Goldschmidt H, Niesvizky R, Joshua D, Chng WJ, Oriol A, et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:1327–37.
Watanabe T, Tobinai K, Matsumoto M, Suzuki K, Sunami K, Ishida T, et al. A phase 1/2 study of carfilzomib in Japanese patients with relapsed and/or refractory multiple myeloma. Br J Haematol. 2016;172:745–56.
Iida S, Watanabe T, Matsumoto M, Suzuki K, Sunami K, Ishida T, et al. Carfilzomib monotherapy in Japanese patients with relapsed or refractory multiple myeloma: a phase 1/2 study. Cancer Sci. 2019;110:2924–32.
Suzuki K, Ri M, Chou T, Sugiura I, Yakezako N, Sunami K, et al. Carfilzomib, lenalidomide and dexamethasone in patients with heavily pretreated multiple myeloma: a phase 1 study in Japan. Cancer Sci. 2017;108:461–8.
Iida S, Tobinai K, Taniwaki M, Shumiya Y, Nakamura T, Chou T. Phase I dose escalation study of high dose carfilzomib monotherapy for Japanese patients with relapsed or refractory multiple myeloma. Int J Hematol. 2016;104:596–604.
Dimopoulos MA, Stewart AK, Masszi T, Špička I, Oriol A, Hájek R, et al. Carfilzomib, lenalidomide, and dexamethasone in patients with relapsed multiple myeloma categorised by age: secondary analysis from the phase 3 ASPIRE study. Br J Haematol. 2017;177:404–13.
Dimopoulos MA, Stewart AK, Masszi T, Špička I, Oriol A, Hájek R, et al. Carfilzomib-lenalidomide-dexamethasone vs lenalidomide-dexamethasone in relapsed multiple myeloma by previous treatment. Blood Cancer J. 2017;7:e554.
Moreau P, Masszi T, Grzasko N, Bahlis NJ, Hansson M, Pour L, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374:1621–34.
Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621–31.
Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319–31.
Suzuki K, Sunami K, Ohashi K, Iida S, Mori T, Handa H, et al. Randomized phase 3 study of elotuzumab for relapsed or refractory multiple myeloma: ELOQUENT-2 Japanese patient subanalysis. Blood Cancer J. 2017;7:e540.
Richardson PG, Xie W, Jagannath S, Jakubowiak A, Lonial S, Raje NS, et al. A phase 2 trial of lenalidomide, bortezomib, and dexamethasone in patients with relapsed and relapsed/refractory myeloma. Blood. 2014;123:1461–9.
Jakubowiak AJ, Dytfeld D, Griffith KA, Lebovic D, Vesole DH, Jagannath S, et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood. 2012;120:1801–9.
Dytfeld D, Jasielec J, Griffith KA, Lebovic D, Vesole DH, Jagannath S, et al. Carfilzomib, lenalidomide, and low-dose dexamethasone in elderly patients with newly diagnosed multiple myeloma. Haematologica. 2014;99:e162–4.
Korde N, Roschewski M, Zingone A, Kwok M, Manasanch EE, Bhutani M, et al. Treatment with carfilzomib-lenalidomide-dexamethasone with lenalidomide extension in patients with smoldering or newly diagnosed multiple myeloma. JAMA Oncol. 2015;1:746–54.
Acknowledgements
We thank all study participants and their families. We thank all study sites and investigators. We would also like to thank the medical consultant, Dr. Hirokazu Murakami (Gunma University Graduate School of Health Science, Maebashi), and Dr. Chihiro Shimazaki (Japan Community Health Care Organization Kyoto-Kuramaguchi Medical Center, Kyoto), Dr. Masahiro Kizaki (Saitama Medical Center, Saitama Medical University, Saitama), Dr. Takao Katoh (International University of Health and Welfare, Mita Hospital, Tokyo), Dr. Masahiro Endo (Shizuoka Cancer Center, Nagaizumi), and Dr. Terufumi Kato (Kanagawa Cancer Center, Yokohama) for their review of the clinical data as members of the Efficacy and Safety Evaluation Committee. We also acknowledge the statistical support of Naokazu Gion and Toshiaki Ozaki (Ono Pharmaceutical, Osaka) and the critical review of the manuscript by Amgen (Thousand Oaks). We thank Susan Cottrell, PhD, and Keyra Martinez Dunn, MD, of Edanz Medical Writing for providing medical writing support, which was funded by Ono Pharmaceutical through EMC K.K. in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Funding
IS, KS, MR, TC, NT, KS, T Ishida, T Izumi, SO, and SI received grants from Ono Pharmaceutical during the course of this study. MR also received grants and personal fees from Bristol-Myers Squibb, Celgene, Janssen Pharmaceutical, Kyowa Hakko Kirin, Ono Pharmaceutical, and Takeda Pharmaceutical, outside the submitted work, and personal fees from Novartis and Sanofi, outside the submitted work. TC also received personal fees from Ono Pharmaceutical during the course of the study and personal fees from Janssen Pharmaceutical, Ono Pharmaceutical, and Takeda Pharmaceutical, outside the submitted work. NT also received personal fees from Ono Pharmaceutical during the course of this study. KS also received personal fees from Ono Pharmaceutical during the course of this study; grants from AbbVie, Alexion Pharma, Daiichi Sankyo, GlaxoSmithKline, Janssen Pharmaceutical, MSD, Novartis, and Sanofi, outside the submitted work; and grants and personal fees from Bristol-Myers Squibb, Celgene, and Takeda Pharmaceutical, outside the submitted work. T Ishida also received personal fees from Celgene, Janssen Pharmaceutical, Ono Pharmaceutical, and Takeda Pharmaceutical, outside the submitted work. SI also received personal fees from Ono Pharmaceutical during the course of this study; grants and personal fees from Bristol-Myers Squibb, Celgene, Janssen Pharmaceutical, Novartis, and Takeda Pharmaceutical, outside the submitted work; and grants from AbbVie, Chugai Pharmaceutical, Daiichi Sankyo, Gilead Pharmaceutical, Kyowa Hakko Kirin, MSD, and Sanofi, outside the submitted work. YS is an employee of Ono Pharmaceutical. The study was designed under the responsibility of Ono Pharmaceutical, in conjunction with the steering committee; the study was funded by Ono Pharmaceutical; carfilzomib was provided by Ono Pharmaceutical; Ono Pharmaceutical collected and analyzed the data and contributed to the interpretation of the study. All authors had full access to all of the data in the study and had final responsibility for the decision to submit for publication.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Sugiura, I., Suzuki, K., Ri, M. et al. Final results of a phase I study of carfilzomib, lenalidomide, and dexamethasone for heavily pretreated multiple myeloma. Int J Hematol 111, 57–64 (2020). https://doi.org/10.1007/s12185-019-02754-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-019-02754-3