
Abstract
Thrombotic thrombocytopenic purpura (TTP) can rapidly progress into a life-threatening condition, thus the importance of appropriate diagnosis and treatment cannot be overstated. Until recently, TTP has mainly been diagnosed by clinical findings such as thrombocytopenia and non-immune hemolytic anemia. In addition to these clinical findings, however, reduced activity of a disintegrin-like and metalloprotease with thrombospondin type 1 motif 13 (ADAMTS13) below 10% has been accepted internationally as a diagnostic criterion for TTP. In the present guidelines, we have taken all of these criteria into consideration. TTP is classified as acquired if the patient is positive for anti-ADAMTS13 autoantibodies, and as congenital if ADAMTS13 gene abnormalities are detected. Fresh-frozen plasma (FFP) transfusion is performed in patients with congenital TTP to supplement ADAMTS13. Plasma exchange therapy using FFP is conducted in patients with acquired TTP to supplement ADAMTS13 and remove anti-ADAMTS13 autoantibodies. To suppress autoantibody production, corticosteroid therapy may be administered in conjunction with plasma exchange. Recent reports show that the monoclonal anti-CD-20 antibody rituximab is effective in patients with refractory or relapsed TTP.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thrombotic thrombocytopenic purpura (TTP) is a serious condition that develops as a result of platelet aggregation and systemic microvascular thrombosis [1]. Along with hemolytic uremic syndrome (HUS), TTP is a typical example of a thrombotic microangiopathy (TMA). TTP is characterized as either acquired or congenital. Previously, due to the absence of specific diagnostic markers, TTP was diagnosed primarily on the basis of the classic pentad of five signs and symptoms: thrombocytopenia, hemolytic anemia, renal failure, fever, and neurologic deficits. However, recent studies demonstrated an association between pathogenesis and severely reduced activity of a disintegrin-like and metalloprotease with thrombospondin type 1 motif 13 (ADAMTS13) [2, 3]. This enzyme specifically cleaves von Willebrand factor (VWF), a glycoprotein necessary for normal hemostasis. Consequently, reduced ADAMTS13 activity has been included in the diagnostic criteria for TTP [4]. Congenital TTP is attributable to ADAMTS13 gene mutations [5, 6], whereas acquired TTP develops as a result of anti-ADAMTS13 autoantibody production [2, 3].
The present diagnostic and treatment guidelines represent the consensus of expert physicians who are members of the TTP group in the Blood Coagulation Abnormalities team in the Health, Labour, and Welfare Scientific Research Grant Project funded by the Japanese government. Given the rarity of this disease, the authors acknowledge the difficulty of reviewing large populations of TTP patients. Nonetheless, we seek to provide the best possible evidence-based description of this condition. In these guidelines, separate sections are devoted to acquired and congenital TTP, and treatment recommendations are sorted by different levels of evidence, following the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE) system (Table 1). Treatments for acquired TTP relate only to adult patients, and attention should be paid to pharmacotherapies for pediatric TTP patients due to the lack of clinical experience in this population. Patients whose ADAMTS13 activity is ≥10% fall outside the scope of these guidelines, because such patients do not meet the criteria for designated intractable/rare diseases made by Japanese Ministry of Health, Labour and Welfare (MHLW) (ADAMTS13 activity <10% for TTP). That said, a certain population of patients clinically diagnosed with TTP have been confirmed to show ADAMTS13 activity above 10%. Further research must be done to better understand their pathology. As with TTP patients who meet the criteria of the present guidelines, a proportion of such patients should undergo plasma exchange and other appropriate treatment without delay.
Pathology
ADAMTS13 is the 13th member of the ADAMTS family of metalloproteases [7]. VWF, the substrate of ADAMTS13, is mainly produced in vascular endothelial cells and released into the plasma as unusually large multimers, which readily undergo ADAMTS13 proteolysis [1]. The platelet-binding affinity of VWF multimers is dependent on their molecular weight. Thus, unusually large VWF multimers are more likely to form platelet thrombi than normal VWF [8]. Platelet–VWF interaction and adhesion are facilitated by high shear stress [9], which develops in the presence of fast blood flow and in small vessels. In patients with TTP, endothelial unusually large VWF multimers secreted into the blood stream tend to remain uncleaved due to low ADAMTS13 activity. In microvessels, exposure to high fluid shear stress induces a conformational change in VWF, which leads to platelet adhesion and subsequent thrombosis. Platelet thrombosis in the microvasculature causes damage to the kidneys, brain, and other end organs.
ADAMTS13 test
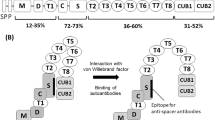
Initially, ADAMTS13 proteolytic activity was measured using full-length VWF as a substrate [10, 11]. Because this method is time-consuming, recent assays employ shorter synthetic derivatives [12,13,14], yielding results in several hours. Results are commonly reported relative to local pooled normal plasmas that are arbitrarily assigned values in units or percentages (1 U/mL or 100% is equivalent to the pooled normal plasma concentration of ADAMTS13).
Two types of anti-ADAMTS13 autoantibodies are recognized: neutralizing and non-neutralizing [15]. The former inhibits the protease in vitro (inhibitor), whereas the latter binds to, but does not suppress it. The majority of anti-ADAMTS13 autoantibodies are inhibitory immunoglobulin G (IgG), although a certain proportion of them are IgA or IgM [16]. In routine clinical practice, it is common to perform assays for only inhibitory ADAMTS13 autoantibodies, and non-neutralizing antibodies are measured largely for research purposes. One Bethesda unit (BU) is defined on the basis of 50% inhibition of ADAMTS13 activity when the test plasma is mixed with an equal amount of normal plasma [17]. The majority of patients with acquired TTP have demonstrable levels of inhibitors [18, 19]. However, low inhibitor titers <1 BU/mL do not constitute convincing evidence for the diagnosis of acquired TTP. As described below, patients who are negative for ADAMTS13 inhibitors may nonetheless have acquired TTP involving autoantibody-mediated ADAMTS13 deficiency. Such patients have non-neutralizing autoantibodies, the presence of which is implicated by their clinical manifestations.
As of the end of March 2017, laboratory tests for ADAMTS13 activity and inhibitory autoantibodies are not subject to reimbursement in Japan.
Acquired TTP
Historical background
In 1924, the American physician Eli Moschcowitz documented the first presumed case of acquired TTP [20], and a series of similar cases without clear etiology were reported thereafter. In 1966, Amorosi and Ultmann described five common signs and symptoms [21], which are referred to as the classic TTP pentad. Among them, the diagnostic importance of thrombocytopenia and hemolytic anemia have gained increasing attention, and these conditions underscore the utility of plasma exchange [22]. Singer and colleagues were the first to use the term TTP, in a 1947 publication [23].
Whereas the clinical benefit of plasma exchange for treating TTP was demonstrated in 1991, its pathogenetic mechanism remained unknown. In the early 1980s, findings of unusually large VWF multimers in the plasma of TTP patients [24] and VWF-rich platelet thrombi at autopsy [25] suggested the involvement of VWF. In 1996, the assay method for VWF protease (ADAMTS13) activity was established [10, 11], and patients with TTP were reported in 1998 to show severely reduced ADAMTS13 activity [2, 3]. However, given that the diagnosis of acquired TTP is not necessarily associated with a marked reduction in ADAMTS13 activity [26,27,28], ADAMTS13 deficiency and clinical manifestations remained distinct diagnostic criteria. Physicians have debated the diagnostic threshold for ADAMTS13 activity reduction. There is growing international consensus on the use of activity levels <10% to support the diagnosis of TTP [29].
Definition
This condition is caused by systemic microvascular aggregation of platelets as a result of autoantibody-mediated severe ADAMTS13 deficiency.
Epidemiology
Overseas, the annual incidence rate of clinically diagnosed TTP has been estimated at 4–11 per million population [30, 31]. The incidence based on ADAMTS13 measurement is unknown. One group of researchers reported an incidence of 1.74 per million population using a diagnostic criterion of ADAMTS13 activity <5% [30]. In overseas studies of TTP based on ADAMTS13 activity, the median age at onset ranged from 36 to 51 years, with the proportion of females ranging from 65 to 100% [18, 27, 32, 33]. In a large study of Japanese patients with ADAMTS13-deficient TTP (ADAMTS13 activity <5%), median age at onset was 54 years, with a female proportion of 55% [19].
Diagnostic algorithm
If a patient presents with reduced platelet count and hemolytic anemia of unknown cause, the clinician should measure ADAMTS13 activity (Fig. 1). If the activity level is <10%, the patient is diagnosed with TTP. If the patient is positive for anti-ADAMTS13 autoantibodies, the patient is diagnosed with acquired TTP. Clinicians should be aware that patients may present with anti-ADAMTS13 autoantibodies even if they are negative for inhibitory autoantibodies. If the patient has no underlying disorders, they are diagnosed with primary acquired TTP. If anti-ADAMTS13 autoantibodies are produced secondary to systemic lupus erythematosus or other autoimmune diseases, or in response to ticlopidine or similar drugs, the diagnosis is secondary acquired TTP.

Diagnostic and treatment algorithm for TMA. (Asterisk) The population of patients with acquired TTP includes a group of patients who are negative for ADAMTS13 inhibitors but positive for anti-ADAMTS13 antibodies. (Double asterisks) The term of atypical HUS (aHUS) is sometimes employed conventionally in health insurance and other documentation. TMA thrombotic microangiopathy, TTP thrombotic thrombocytopenic purpura, ADAMTS13 a disintegrin-like and metalloprotease with thrombospondin type 1 motif 13, aHUS atypical hemolytic uremic syndrome, FFP fresh frozen plasma, STEC Shiga toxin-producing Escherichia coli
General laboratory thresholds related to the classic TTP pentad
Thrombocytopenia
The general upper limit for platelet count is 100,000/μL for the diagnosis of thrombocytopenia. Most patients with TTP have platelet counts in the range of 10,000–30,000/μL. The median platelet count in a cohort of Japanese patients with TTP was 10,000/μL [19].
Hemolytic anemia
Patients with acquired TTP exhibit microangiopathic hemolytic anemia, characterized by mechanical damage to red blood cells. Hemoglobin levels are typically in the range of 8–10 g/dL in overseas patients with TTP [34], whereas the median value in Japanese TTP patients has been reported as 7.3 g/dL [19]. The diagnosis of hemolytic anemia is made based on the following: the presence of schistocytes; increased levels of indirect bilirubin, lactate dehydrogenase, and reticulocytes; significantly decreased levels of haptoglobin; and negative direct Coombs test. Peripheral smears showing schistocyte percentage >1% in the absence of additional severe erythrocyte shape abnormalities are recommended as a robust indication for the diagnosis of TMA [35]. However, clinicians should be careful in regarding schistocytes as the hallmark of the disease, as the quantification of morphologic changes of blood cells is technically challenging and schistocytes may not always be present in the blood samples collected from patients with TTP.
Renal impairment
Renal impairment may present as mild changes involving occult blood or positive urinary protein alone, while more severe cases may involve elevated serum creatinine levels. In most cases, the serum creatinine levels are <2 mg/dL [19]. Severe acute renal dysfunction that requires immediate hemodialysis is suggestive of HUS.
Fever
Patients with TTP may present with a mild (37–39 °C) or high fever (>39 °C). Fever is documented in approximately 30% [32, 33] to 72% [19] of patients with TTP.
Fluctuating neurologic conditions
Patients with TTP may develop a variety of neurologic conditions, ranging from mild headache to more severe manifestations involving delirium, confusion, personality change, decreased level of consciousness, quadriplegia, convulsions, etc. Neurologic manifestations usually fluctuate in severity and location, and often improve dramatically following plasma exchange. Neurologic involvement was reported in 79% of Japanese patients [19], and several overseas studies reported a rate of approximately 50% [18, 27].
Differential diagnosis
The diagnostic criterion based on ADAMTS13 activity provides a clear guidance for differentiating several common disorders from TTP, as described below. TMAs are subclassified by etiology. These etiology-based subclassifications correspond to clinical diagnoses, as shown in Table 2.
Disseminated intravascular coagulation
In patients with TTP, prothrombin time (PT) and activated partial thromboplastin time (APTT) are normal, fibrinogen and antithrombin levels typically remain in the normal range, and fibrin/fibrinogen degradation products (FDP) and d-dimer levels are mildly elevated. In contrast, disseminated intravascular coagulation (DIC) is usually characterized by fibrin thrombi, prolonged PT and APTT times, and reduced levels of fibrinogen.
Hemolytic uremic syndrome
HUS induced by Shiga toxin-producing Escherichia coli (STEC) strains, such as E. coli O157, can be diagnosed using (a) stool culture, enzyme immunoassay (EIA), and other methods for detecting STEC, or (b) methods for detecting anti-lipopolysaccharide (LPS) IgM antibodies. Formerly, cases of HUS unrelated to STEC infection were referred to as atypical HUS (aHUS). More recently, the term aHUS has been used to refer only to HUS involving complement abnormalities, such as complement factor H, component 3 (C3), and other elements [36], and is classified etiologically as a complement-mediated TMA.
HELLP syndrome
HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome occurs in connection with pregnancy-induced hypertensive nephropathy or preeclampsia, and can lead to multiple organ failure. The diagnostic criteria proposed by Sibai et al. are commonly used [37], but these criteria are not helpful for discriminating between HELLP syndrome and TTP. Patients with severely reduced ADAMTS13 activity should be diagnosed with TTP.
Evans syndrome
Evans syndrome is defined as the combination of autoimmune hemolytic anemia and idiopathic thrombocytopenic purpura (ITP). Patients with Evans syndrome demonstrate a positive direct Coombs test. Certain patients diagnosed with Evans syndrome are direct Coombs-negative, and this patient group may include those who should be diagnosed with TTP based on a severe decrease in ADAMTS13 activity.
Secondary thrombotic microangiopathy
TMAs are reported to develop secondary to the following: systemic lupus erythematosus, scleroderma, and other connective tissue diseases; transplantation of hematopoietic stem cells, kidneys, and other organs; and malignant diseases. Although the exact etiology of secondary TMA is unknown, many scientists believe that it is primarily attributable to endothelial cell injury.
Other thrombotic microangiopathies
Some patients who are clinically diagnosed with TMA in the absence of underlying conditions do not satisfy the TTP criterion of a severe decrease of ADAMTS13 activity to <10%. This group of patients includes those who were formerly diagnosed with TTP based on the classic pentad.
Treatments
Plasma exchange is the only scientifically established treatment modality for acquired TTP [22]. In light of a report indicating that longer delay in initiating plasma exchange is an independent predictor of treatment failure [38], the clinician should initiate plasma exchange as soon as possible if the patient is suspected of having acquired TTP. The clinician should implement plasma exchange before the ADAMTS13 assay report is ready, since final results may not be available for several days after the assay is ordered. The following describes current treatment recommendations, along with grades of the strength of the recommendation (Table 1).
Acute phase
-
a)
Plasma exchange (grade of recommendation: 1A)
The physician should carry out plasma exchange with FFP once daily. The FFP volume is 1.0–1.5 times of the patient’s circulating plasma volume, which can be estimated using the equation below. The effectiveness of plasma exchange is attributable to: (1) ADAMTS13 supplementation, (2) removal of ADAMTS13 inhibitors, and (3) elimination of unusually large VWF multimers unsusceptible to proteolytic cleavage [28]. Human serum albumin solutions are not recommended for replacement, because these do not supplement ADAMTS13. FFP infusion may be performed in emergency cases. An analysis of response and survival showed that plasma exchange is superior to plasma infusion in the treatment of TTP [22].
Circulating plasma volume (mL) = body weight (kg) × 70 (mL/kg) × (1 − hematocrit value [%]/100).
Plasma exchange
The physician should perform plasma exchange with FFP (50–75 mL/kg) once daily. Ideally, plasma exchange should be performed daily until 2 days after the platelet count is normalized (≥150,000/μL), as recommended by the UK guideline group [34]. However, this may not be practical in Japan due to Japanese health insurance policy restrictions (i.e., ≤3 times/week for a maximum of 3 months). For this reason, the physician should perform plasma exchange once daily for the first 5 days, followed by administration once every other day. The physician should determine the plasma exchange schedule based on the time course of change in platelet count and ADAMTS13 test results. Critically ill TTP patients may require plasma exchange more frequently than the government-determined reimbursable upper limit of 12 times per month.
-
b)
Corticosteroid therapy (grade of recommendation: 1B)
Corticosteroids are administered either as intravenous pulse therapy or high-dose oral therapy. Available data on the superiority of either modality are inconclusive [39]. Corticosteroids are expected to suppress autoantibody production. The physician should consider lower doses in patients who are elderly or who have diabetes mellitus or severe infections. Although corticosteroid use in patients with TTP is considered off-label under current Japanese health insurance standards, it is classified as a Grade-1 recommendation based on its popularity as a TTP treatment in routine clinical practice.
Pulse corticosteroid therapy (not covered by Japanese health insurance).
Methylprednisolone 1000 mg/day for 3 days.
After receiving plasma exchange, the patient is administered methylprednisolone 1000 mg once daily over approximately 2 h by drip infusion. After the corticosteroid is administered by drip infusion at 1000 mg/day for three consecutive days, the dose should be tapered. The dose reduction regimen should be made with reference to platelet count and ADAMTS13 test results. No evidence-based procedure has been established for corticosteroid dose reduction. If the physician wishes to extend methylprednisolone therapy for the treatment of neurologic symptoms, or under conditions of intensive care unit (ICU) management, the corticosteroid may be administered at 500, 250, and 125 mg/day for 2 days each in this order. The dosage regimen should then be switched to oral corticosteroids 30 mg/day, and subsequently tapered with reference to the descriptions below. If the physician decides to switch to oral therapy after the first 3 days of drip infusion, the physician should orally administer predonisolone 0.5–1.0 mg/kg/day and taper the dose based on the following descriptions:
High-dose oral corticosteroid therapy (not covered by Japanese health insurance).
Prednisolone 1 mg/kg/day.
The physician tapers the prednisolone dose with reference to the platelet count and ADAMTS13 test results. The initial dose (1 mg/kg/day) is maintained for 2 weeks, followed by a rapid reduction to 0.5 mg/kg/day. The physician should further reduce the dose to the range of approximately 2.5–5.0 mg/week, taking note of the platelet count and ADAMTS13 test results.
-
c)
Antiplatelet drugs (grade of recommendation: 2B)
Aspirin may be administered when the platelet count recovers to >50,000/μL. However, its impact on preventing TTP relapse is unsubstantiated.
Antiplatelet pharmacotherapy (not covered by Japanese health insurance).
Oral aspirin may be administered at a dose range of 81–100 mg once daily in the morning until the termination of corticosteroid use.
-
d)
Other treatments
Patients without cardiac disorders may undergo red blood cell transfusion if hemoglobin levels decrease to <7.0 g/dL [34, 40]. In patients with cardiac disorders, hemoglobin levels <8.0 g/dL may suggest the need for red blood cell transfusion (grade of recommendation: 1A) [41]. Platelet transfusion may be indicated for patients with life-threatening bleeding. Prophylactic platelet transfusion in other situations, however, is contraindicated, as it may aggravate thrombosis (grade of recommendation: 1B) [34, 42, 43].
In patients manifesting TTP secondary to the use of a specific drug, the drug should be immediately discontinued. If the patient has an underlying condition, only drugs used to treat the condition should be continued. The physician conducts plasma exchange in patients with secondary TTP in a manner similar to those with primary TTP, as their ADAMTS13 activity levels may be severely lowered.
Patients with refractory TTP and those with early relapse
If the platelet count does not increase to 50,000/μL after five plasma exchange sessions, or if the platelet count decreases to <50,000/μL after initially recovering to >150,000/μL, the physician should consider rituximab in combination with plasma exchange (grade of recommendation: 1B) [44]. The physician should preferably order both an ADAMTS13 activity assay and an inhibitor screen, as repeated plasma exchange sessions may elevate ADAMTS13 inhibitor titers [45]. Patients with a boosted inhibitor titer are often unresponsive to plasma exchange alone, thus necessitating combined use of rituximab. Rituximab takes 10–14 days to demonstrate a clinical impact, and plasma exchange should be conducted in the interim.
-
a)
Rituximab (grade of recommendation: 1B) (not covered by Japanese health insurance)
Rituximab, an anti-CD20 monoclonal antibody, suppresses ADAMTS13 inhibitor production by depleting B lymphocytes. Although rituximab therapy for the treatment of TTP is not covered by Japanese health insurance, it is classified as Grade 1, as it is widely administered in clinical practice for treating patients with refractory TTP [44, 46, 47].
Rituximab therapy.
Rituximab 375 mg/m2.
Rituximab is administered at increasing infusion rates using an infusion pump. It is administered after plasma exchange where appropriate.
The recommended rituximab therapy regimen is once weekly for four doses.
In addition to rituximab, the following therapies are typically available, but not always effective, for treating refractory or recurrent cases:
-
b)
Cyclophosphamide (grade of recommendation: 2B) (not covered by Japanese health insurance) [48]
Cyclophosphamide 500 mg/body.
Cyclophosphamide is administered once daily over 2 h. Typically, the cyclophosphamide regimen is restricted to a single dose, as multiple doses may cause bone marrow suppression.
-
c)
Vincristine (grade of recommendation: 2B) (not covered by Japanese health insurance) [49]
Vincristine 1 mg/body is administered intravenously once daily at a slow rate of infusion. The vincristine regimen is usually restricted to a single dose, as multiple doses may cause neurotoxicity and bone marrow suppression.
-
d)
Cyclosporine (grade of recommendation: 2B) (not covered by Japanese health insurance) [50]
Oral cyclosporine 4 mg/kg/day is administered in two divided doses. Blood levels of cyclosporine should be monitored to maintain a trough level of approximately 100–200 ng/mL.
-
e)
Other treatments (not covered by Japanese health insurance)
Formerly, splenectomy (grade of recommendation: 2C) [51] and high-dose immunoglobulin (grade of recommendation: 2C) [52] were frequently administered to treat patients with refractory or recurrent TTP. These treatments have been superseded by rituximab therapy.
Remission period
After the patient has achieved full remission, the physician should discontinue corticosteroids at the earliest opportunity, based on ADAMTS13 activity and inhibitor titer measurements. No effective prophylactic treatments are recommended for patients in remission. To minimize the risk of relapse, patients should visit the physician regularly during the first several years after achieving remission and undergo monitoring of the platelet count and ADAMTS13 activity. In patients in remission with normal platelet counts, ADAMTS13 activity may be severely reduced and ADAMTS13 inhibitors may be present [16, 53]. Patients whose ADAMTS13 activity is significantly reduced or who are positive for ADAMTS13 inhibitors are at high risk of relapse [16, 53].
Classification by severity
Table 3 summarizes the TTP severity classification scheme used in designated intractable/rare diseases made by Japanese MHLW for a medical expense support system. If the patient’s condition meets one or more of these eight criteria, the condition is classified as moderate or severe, and the patient is eligible for the designated intractable/rare disease program.
Treatment outcome
TTP was formerly associated with a poor prognosis, with a mortality rate higher than 90% in untreated patients [21]. The introduction of plasma exchange, however, has elevated the survival rate to approximately 80% [18, 19, 22, 27, 32]. High levels of serum creatinine and ADAMTS13 inhibitor titers of two or more BU/mL are poor prognostic factors for patients with ADAMTS13 activities <10% [18]. Patients with acute TTP are at risk of cardiovascular death, and should undergo myocardial troponin I measurement [34].
Congenital TTP (Upshaw-Schulman syndrome)
Historical background
Early in the 1950s, neonatal cases characterized by severe jaundice and thrombocytopenia were documented [54]. In 1960, Schuman and colleagues [55] reported an 8-year-old girl who experienced repeated bleeding events associated with chronic thrombocytopenia from the neonatal period. One key clinical characteristic of this patient was that her platelet count remarkably improved following transfusion of small amounts of FFP. In 1978, Upshaw reported a similar case of a 28-year-old woman with chronic thrombocytopenia who also responded well to FFP transfusion [56]. These conditions were referred to as USS [57], and low blood fibronectin levels were associated with its etiology, although this theory was later refuted. Thereafter, the term USS was abandoned in the Western medical community and superseded by “chronic-relapsing (CR) TTP” [24]. While the latter term gained lasting popularity, it obscured the fact that TTP can be differentiated into congenital and acquired types. In 1982, Moake et al. made the important discovery that unusually large VWF multimers appear in patients with remitted CR-TTP [24, 58]. In 1997, Furlan et al. reported that VWF-cleaving protease (CP) activity is severely reduced in patients with CR-TTP [58]. The VWF-CP was later identified as ADAMTS13. The CR-TTP patient cohorts in these studies included individuals with both congenital and acquired TTP. Furlan et al. reported that the parents of the two sibling patients in the study had essentially normal VWF-CP activity. In a study of three Japanese USS patients reported in July 2001 [59], Kinoshita et al. showed that the VWF-CP activities of the patients were undetectable, while those of their parents ranged from 5.6 to 60% of control values. The authors concluded that USS is inherited in an autosomal recessive fashion. Levy et al. conducted genome-wide positional cloning in four pedigrees of individuals with congenital TTP, and reported in an article published in December 2001 that ADAMTS13 was the responsible gene [5]. These findings led to the recognition that USS is a congenital form of TTP arising from ADAMTS13 gene abnormalities, and thus the terms “USS” and “congenital TTP” are synonyms. Two simple assays for determining ADAMTS13 activity were recently developed in Japan [13, 14], and these have accelerated the diagnosis of USS patients. At present, approximately one-third of the USS patients identified worldwide are from Japan.
Definition
Congenital TTP is a hereditary disorder resulting from systemic microvascular aggregation of platelets, and is attributable to ADAMTS13 gene mutations causing severe ADAMTS13 deficiency. Its inheritance is autosomal recessive.
Epidemiology
The prevalence rate of congenital TTP is unknown. The population of patients with congenital TTP is assumed to be much smaller than that of patients with acquired TTP [60]. One European registry-based study estimated that the prevalence was in the range of 0.5–4 per million population [61]. A group of Japanese researchers estimated that this disease develops in 1 out of 1.1 million population [62]. By the end of 2015, 59 patients with congenital TTP have been identified in Japan. Since this disease exhibits autosomal recessive inheritance, it should theoretically develop at equal rates in both sexes. However, the group of 59 patients documented in Japan consists of 24 males and 35 females. The larger number of female patients with congenital TTP may be attributable to the greater chance of detection during pregnancy.
Depending on the timing of onset, this disease can be subcategorized into two types [63]:
Early onset Typically, patients with early-onset congenital TTP have a history of replacement transfusion for the treatment of severe neonatal jaundice. After repeated thrombocytopenic episodes in association with influenza and other infections, patients are diagnosed with congenital TTP and receive periodic FFP transfusion during childhood.
Adult onset Patients are diagnosed with congenital TTP in adulthood after experiencing thrombocytopenia in association with pregnancy, influenza, or other infections. Some patients who experience the onset of TTP in adulthood may have been misdiagnosed with ITP due to childhood thrombocytopenic episodes. As patients with congenital TTP have mild hemolytic anemia, they may remain undetected and undiagnosed. In female patients with congenital TTP, pregnancy will provide a unique opportunity for detecting TTP signs and symptoms [64]. One Japanese male patient was diagnosed with TTP for the first time at the age of 63 [65].
Diagnosis
Congenital TTP is suspected if the patient presents with thrombocytopenia and ADAMTS13 activity <10% in the absence of ADAMTS13 inhibitors. However, it is not always technically feasible to determine whether the patient is positive or negative for ADAMTS13 inhibitors. In certain situations, differentiation between congenital and acquired TTP can only be made after monitoring ADAMTS13 activity and testing the ADAMTS13 activity of their parents. ADAMTS13 gene analysis is required for the definitive diagnosis of congenital TTP. Because asymptomatic parents of a patient with congenital TTP have heterozygous mutations, their ADAMTS13 activities often range from 30 to 50% [6, 66].
Congenital TTP should be differentiated from ITP unresponsive to common medical management, as well as from pregnancy-induced complement-mediated TMA and HELLP syndrome. The physician should note that although congenital TTP always involves severely decreased ADAMTS13 activity, it is not necessarily accompanied by thrombocytopenia.
ADAMTS13 gene analysis
Congenital TTP is an autosomal recessive disorder involving homozygous or compound heterozygous ADAMTS13 gene mutations [5, 67]. Of 55 Japanese patients with genetically confirmed congenital TTP, 10 and 45 had homozygous and compound heterozygous ADAMTS13 gene mutations, respectively.
Treatment
FFP transfusion regimens effective in patients with congenital TTP vary depending on their condition; some patients require chronic treatment with periodic FFP transfusions, while others require episodic FFP transfusions only when their condition worsens. The volume of FFP and the duration of transfusion should be determined by taking into consideration platelet count changes and occult blood scores.
Fresh frozen plasma transfusion (grade of recommendation: 1B).
The physician should administer FFP at a volume of 5–10 mL/kg once every 2–3 weeks. At the time of onset, the physician should transfuse FFP at a volume of 10 mL/kg and monitor the clinical impact. To minimize the risks of allergy/anaphylaxis, disease transmission, and other adverse reactions to FFP infusion at the time of onset, efforts should be made to collect FFP from the smallest possible number of donors.
Classification by severity
Table 4 presents the congenital TTP severity classification scheme used in designated intractable/rare diseases made by Japanese MHLW for a medical expense support system. All cases of congenital TTP are covered by this system, except for mild cases that require no medical treatment.
Treatment outcome
Appropriate diagnosis and treatment with FFP transfusion will generally achieve excellent results in patients with congenital TTP. In Japan, eight deaths were documented among 59 patients diagnosed with congenital TTP. Five of these died after the start of hemodialysis treatment. These cases suggest that preventing renal deterioration may improve outcomes. It has been suggested that when FFP administration leads to anti-ADAMTS13 alloantibody formation in patients with congenital TTP, the clinical effects of FFP will be compromised. Although no such cases have been reported to date in Japan, this possibility underscores the importance of performing regular ADAMTS13 inhibitor assays.
References
Moake JL. Thrombotic thrombocytopenic purpura: the systemic clumping “plague”. Annu Rev Med. 2002;53:75–88.
Furlan M, Robles R, Galbusera M, Remuzzi G, Kyrle PA, Brenner B, et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339:1578–84.
Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339:1585–94.
George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371:1847–8.
Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413:488–94.
Kokame K, Matsumoto M, Soejima K, Yagi H, Ishizashi H, Funato M, et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc Natl Acad Sci USA. 2002;99:11902–7.
Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276:41059–63.
Von Furlan M. Willebrand factor: molecular size and functional activity. Ann Hematol. 1996;72:341–8.
Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–97.
Furlan M, Robles R, Lammle B. Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood. 1996;87:4223–34.
Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87:4235–44.
Kokame K, Matsumoto M, Fujimura Y, Miyata T. VWF73, a region from D1596 to R1668 of von Willebrand factor, provides a minimal substrate for ADAMTS-13. Blood. 2004;103:607–12.
Kokame K, Nobe Y, Kokubo Y, Okayama A, Miyata T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005;129:93–100.
Kato S, Matsumoto M, Matsuyama T, Isonishi A, Hiura H, Fujimura Y. Novel monoclonal antibody-based enzyme immunoassay for determining plasma levels of ADAMTS13 activity. Transfusion. 2006;46:1444–52.
Scheiflinger F, Knobl P, Trattner B, Plaimauer B, Mohr G, Dockal M, et al. Nonneutralizing IgM and IgG antibodies to von Willebrand factor-cleaving protease (ADAMTS-13) in a patient with thrombotic thrombocytopenic purpura. Blood. 2003;102:3241–3.
Ferrari S, Scheiflinger F, Rieger M, Mudde G, Wolf M, Coppo P, et al. Prognostic value of anti-ADAMTS 13 antibody features (Ig isotype, titer, and inhibitory effect) in a cohort of 35 adult French patients undergoing a first episode of thrombotic microangiopathy with undetectable ADAMTS 13 activity. Blood. 2007;109:2815–22.
Kasper CK, Pool JG. Letter: measurement of mild factor VIII inhibitors in Bethesda units. Thromb Diath Haemorrh. 1975;34:875–6.
Kremer Hovinga JA, Vesely SK, Terrell DR, Lämmle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115:1500–11.
Matsumoto M, Bennett CL, Isonishi A, Qureshi Z, Hori Y, Hayakawa M, et al. Acquired Idiopathic ADAMTS13 Activity Deficient Thrombotic Thrombocytopenic Purpura in a Population from Japan. PLoS One. 2012;7:e33029.
Moschcowitz E. Hyaline Thrombosis of the terminal arterioles and capillaries: a hitherto undescribed disease. Proc N Y Pathol Soc. 1924;24:21–4.
Amorosi EL, Ultmann JE. Thrombotic thrombocytopenic purpura: report of 16 cases and review of the literature. Medicine. 1966;45:139–59.
Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325:393–7.
Singer K, Bornstein FP, Wile SA. Thrombotic thrombocytopenic purpura; hemorrhagic diathesis with generalized platelet thromboses. Blood. 1947;2:542–54.
Moake JL, Rudy CK, Troll JH, Weinstein MJ, Colannino NM, Azocar J, et al. Unusually large plasma factor VIII: von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. 1982;307:1432–5.
Asada Y, Sumiyoshi A, Hayashi T, Suzumiya J, Kaketani K. Immunohistochemistry of vascular lesion in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb Res. 1985;38:469–79.
Veyradier A, Obert B, Houllier A, Meyer D, Girma JP. Specific von Willebrand factor-cleaving protease in thrombotic microangiopathies: a study of 111 cases. Blood. 2001;98:1765–72.
Vesely SK, George JN, Lämmle B, Studt JD, Alberio L, El-Harake MA, et al. ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients. Blood. 2003;102:60–8.
Matsumoto M, Yagi H, Ishizashi H, Wada H, Fujimura Y. The Japanese experience with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Semin Hematol. 2004;41:68–74.
Scully M, Cataland S, Coppo P, de la Rubia J, Friedman KD, Kremer Hovinga J, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15:312–22.
Terrell DR, Williams LA, Vesely SK, Lammle B, Hovinga JA, George JN. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. J Thromb Haemost. 2005;3:1432–6.
Scully M, Yarranton H, Liesner R, Cavenagh J, Hunt B, Benjamin S, et al. Regional UK TTP registry: correlation with laboratory ADAMTS 13 analysis and clinical features. Br J Haematol. 2008;142:819–26.
Zheng XL, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103:4043–9.
Coppo P, Bengoufa D, Veyradier A, Wolf M, Bussel A, Millot GA, et al. Severe ADAMTS13 deficiency in adult idiopathic thrombotic microangiopathies defines a subset of patients characterized by various autoimmune manifestations, lower platelet count, and mild renal involvement. Medicine (Baltimore). 2004;83:233–44.
Scully M, Hunt BJ, Benjamin S, Liesner R, Rose P, Peyvandi F, et al. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol. 2012;158:323–35.
Zini G, d’Onofrio G, Briggs C, Erber W, Jou JM, Lee SH, et al. ICSH recommendations for identification, diagnostic value, and quantitation of schistocytes. Int J Lab Hematol. 2012;34:107–16.
Kato H, Nangaku M, Hataya H, Sawai T, Ashida A, Fujimaru R, et al. Clinical guides for atypical hemolytic uremic syndrome in Japan. Clin Exp Nephrol. 2016;20:536–43.
Sibai BM, Ramadan MK, Usta I, Salama M, Mercer BM, Friedman SA. Maternal morbidity and mortality in 442 pregnancies with hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome). Am J Obstet Gynecol. 1993;169:1000–6.
Pereira A, Mazzara R, Monteagudo J, Sanz C, Puig L, Martinez A, et al. Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome: a multivariate analysis of factors predicting the response to plasma exchange. Ann Hematol. 1995;70:319–23.
Balduini CL, Gugliotta L, Luppi M, Laurenti L, Klersy C, Pieresca C, et al. High versus standard dose methylprednisolone in the acute phase of idiopathic thrombotic thrombocytopenic purpura: a randomized study. Ann Hematol. 2010;89:591–6.
Holst LB, Haase N, Wetterslev J, Wernerman J, Guttormsen AB, Karlsson S, et al. Lower versus higher hemoglobin threshold for transfusion in septic shock. N Engl J Med. 2014;371:1381–91.
Hebert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med. 1999;340:409–17.
Benhamou Y, Baudel JL, Wynckel A, Galicier L, Azoulay E, Provot F, et al. Are platelet transfusions harmful in acquired thrombotic thrombocytopenic purpura at the acute phase? Experience of the French thrombotic microangiopathies reference center. Am J Hematol. 2015;90:E127–9.
Kumar A, Mhaskar R, Grossman BJ, Kaufman RM, Tobian AA, Kleinman S, et al. Platelet transfusion: a systematic review of the clinical evidence. Transfusion. 2015;55:1116–27.
Miyakawa Y, Imada K, Ichinohe T, Nishio K, Abe T, Murata M, et al. Efficacy and safety of rituximab in Japanese patients with acquired thrombotic thrombocytopenic purpura refractory to conventional therapy. Int J Hematol. 2016;104:228–35.
Isonishi A, Bennett CL, Plaimauer B, Scheiflinger F, Matsumoto M, Fujimura Y. Poor responder to plasma exchange therapy in acquired thrombotic thrombocytopenic purpura is associated with ADAMTS13 inhibitor boosting: visualization of an ADAMTS13 inhibitor complex and its proteolytic clearance from plasma. Transfusion. 2015;55:2321–30.
Scully M, McDonald V, Cavenagh J, Hunt BJ, Longair I, Cohen H, et al. A phase 2 study of the safety and efficacy of rituximab with plasma exchange in acute acquired thrombotic thrombocytopenic purpura. Blood. 2011;118:1746–53.
Froissart A, Buffet M, Veyradier A, Poullin P, Provôt F, Malot S, et al. Efficacy and safety of first-line rituximab in severe, acquired thrombotic thrombocytopenic purpura with a suboptimal response to plasma exchange. Experience of the French Thrombotic Microangiopathies Reference Center. Crit Care Med. 2012;40:104–11.
Beloncle F, Buffet M, Coindre JP, Munoz-Bongrand N, Malot S, Pene F, et al. Splenectomy and/or cyclophosphamide as salvage therapies in thrombotic thrombocytopenic purpura: the French TMA Reference Center experience. Transfusion. 2012;52:2436–44.
Ziman A, Mitri M, Klapper E, Pepkowitz SH, Goldfinger D. Combination vincristine and plasma exchange as initial therapy in patients with thrombotic thrombocytopenic purpura: one institution’s experience and review of the literature. Transfusion. 2005;45:41–9.
Nosari A, Redaelli R, Caimi TM, Mostarda G, Morra E. Cyclosporine therapy in refractory/relapsed patients with thrombotic thrombocytopenic purpura. Am J Hematol. 2009;84:313–4.
Kappers-Klunne MC, Wijermans P, Fijnheer R, Croockewit AJ, van der Holt B, de Wolf JT, et al. Splenectomy for the treatment of thrombotic thrombocytopenic purpura. Br J Haematol. 2005;130:768–76.
Kawano N, Yokota-Ikeda N, Yoshida S, Kuriyama T, Yamashita K, Sugio Y, et al. Therapeutic modality of 11 patients with TTP in a single institution in Miyazaki from 2000 to 2011. Intern Med. 2013;52:1883–91.
Peyvandi F, Lavoretano S, Palla R, Feys HB, Vanhoorelbeke K, Battaglioli T, et al. ADAMTS13 and anti-ADAMTS13 antibodies as markers for recurrence of acquired thrombotic thrombocytopenic purpura during remission. Haematologica. 2008;93:232–9.
Dacie JV, Mollison PL, Richardson N, Selwyn JG, Shapiro L. Atypical congenital haemolytic anaemia. Q J Med. 1953;22:79–98.
Schulman I, Pierce M, Lukens A, Currimbhoy Z. Studies on thrombopoiesis. I. A factor in normal human plasma required for platelet production; chronic thrombocytopenia due to its deficiency. Blood. 1960;16:943–57.
Upshaw JD Jr. Congenital deficiency of a factor in normal plasma that reverses microangiopathic hemolysis and thrombocytopenia. N Engl J Med. 1978;298:1350–2.
Rennard S, Abe S. Decreased cold-insoluble globulin in congenital thrombocytopenia (Upshaw-Schulman syndrome). N Engl J Med. 1979;300:368.
Furlan M, Robles R, Solenthaler M, Wassmer M, Sandoz P, Lammle B. Deficient activity of von Willebrand factor-cleaving protease in chronic relapsing thrombotic thrombocytopenic purpura. Blood. 1997;89:3097–103.
Kinoshita S, Yoshioka A, Park YD, Shizashi H, Konno M, Funato M, et al. Upshaw-Schulman syndrome revisited: a concept of congenital thrombotic thrombocytopenic purpura. Int J Hematol. 2001;74:101–8.
Sadler JE. What’s new in the diagnosis and pathophysiology of thrombotic thrombocytopenic purpura. Hematol Am Soc Hematol Educ Progr. 2015;2015:631–6.
Mansouri Taleghani M, von Krogh AS, Fujimura Y, George JN, Hrachovinova I, Knobl PN, et al. Hereditary thrombotic thrombocytopenic purpura and the hereditary TTP registry. Hamostaseologie. 2013;33:138–43.
Kokame K, Kokubo Y, Miyata T. Polymorphisms and mutations of ADAMTS13 in the Japanese population and estimation of the number of patients with Upshaw-Schulman syndrome. J Thromb Haemost. 2011;9:1654–6.
Fujimura Y, Kokame K, Yagi H, Isonishi A, Matsumoto M, Miyata T. Hereditary deficiency of ADAMTS13 activity: Upshaw-Schulman syndrome. In: Rodgers MD, editor. ADAMTS13. Switzerland: Springer; 2015. p. 73–90.
Fujimura Y, Matsumoto M, Kokame K, Isonishi A, Soejima K, Akiyama N, et al. Pregnancy-induced thrombocytopenia and TTP, and the risk of fetal death, in Upshaw-Schulman syndrome: a series of 15 pregnancies in 9 genotyped patients. Br J Haematol. 2009;144:742–54.
Taguchi F, Yagi H, Matsumoto M, Sadamura S, Isonishi A, Soejima K, et al. The homozygous p. C1024R- ADAMTS13 gene mutation links to a late-onset phenotype of Upshaw-Schulman syndrome in Japan. Thromb Haemost. 2012;107:1003–5.
Matsumoto M, Kokame K, Soejima K, Miura M, Hayashi S, Fujii Y, et al. Molecular characterization of ADAMTS13 gene mutations in Japanese patients with Upshaw-Schulman syndrome. Blood. 2004;103:1305–10.
Fujimura Y, Matsumoto M, Isonishi A, Yagi H, Kokame K, Soejima K, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost. 2011;9(Suppl 1):283–301.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The following authors declare competing interests with respect to this article. Masanori Matsumoto: Received consultant/advisor fees from Baxalta, Alexion Pharma, Ablynx nv and Kainos Laboratories; lecture fees from Asahi Kasei Pharma, Chugai Pharmaceutical, Alexion Pharma, and Bayer Yakuhin; and research funds from Chugai Pharmaceutical, Bayer Yakuhin, and Baxalta. He has shared patent rights with Alfresa Pharma. Yoshihiro Fujimura: Is an employee of the Japanese Red Cross Kinki Block Blood Center. He also has shared patent rights with Alfresa Pharma. Hideo Wada: Received lecture fees from Alexion Pharma and research fund from Daiichi Sankyo. Koichi Kokame: Received patent royalties through National Cerebral and Cardiovascular Center from the Peptide Institute, Peptides International, Kainos Laboratories, SRL, Alfresa Pharma, and AnaSpec. Yoshitaka Miyakawa: Received consultant/advisor fees from Novartis Pharma, Kyowa Hakko Kirin, Kainos Laboratories, and Fujifilm; lecture fees from Novartis Pharma, Kyowa Hakko Kirin, Alexion Pharma, Chugai Pharmaceutical, Bayer Yakuhin, Daiichi Sankyo, Asahi Kasei Pharma, Kissei Pharmaceutical, Biogen, and Baxalta; manuscript fees from Terumo, Zenyaku Kogyo, and the Japan Blood Products Organization; and research funds from Alexion Pharma and Teijin Pharma. Yasunori Ueda: Received consultant/advisor fees from Ablynx nv and Kainos Laboratories; research found for clinical trial from Eli Lilly Japan, Sumitomo Dainippon Pharma, Celgene, Symbio Pharmaceutical, Astellas Pharma, Inc. and Eisai. Takanori Moriki: Is an employee at the Bank of Tokyo-Mitsubishi UFJ, Ltd. Medical Center, Head Office. Toshiyuki Miyata: Received consultant/advisor fees from Alexion Pharma and lecture fees from CSL Behring, LSI Medience, Alexion Pharma, Daiichi Sankyo, and Bayer Yakuhin. He also received patent royalties through National Cerebral and Cardiovascular Center from the Peptide Institute, Peptides International, Kainos Laboratories, SRL, Alfresa Pharma, and AnaSpec. Mitsuru Murata: Received consultant/advisor fees from Pfizer and Otsuka Pharmaceutical; lecture and related fees from Sekisui Medical, LSI Medience, Siemens Healthcare Diagnostics, Sumitomo Dainippon Pharma, Taisho Toyama Pharmaceutical, Tosoh Bioscience, Beckman Coulter, and Alexion Pharma; and research funds from Hitachi High-Technologies, Sanofi, Abbott Japan, Tosoh, and Shino-Test.
Additional information
The Japanese version of these guidelines was published in Jpn J Clin Hematol 2017;58:271–81.
About this article

Cite this article
Matsumoto, M., Fujimura, Y., Wada, H. et al. Diagnostic and treatment guidelines for thrombotic thrombocytopenic purpura (TTP) 2017 in Japan. Int J Hematol 106, 3–15 (2017). https://doi.org/10.1007/s12185-017-2264-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2264-7