
Abstract
Multiple myeloma (MM) is a common hematological malignancy. To identify key genes and microRNAs in MM, we downloaded two gene expression profiles (GSE16558 and GSE47552) and two microRNA expression profiles (GSE17498 and GSE16558) from the Gene Expression Omnibus (GEO) database. A total of 596 differentially expressed genes (DEGs) and 39 differentially expressed microRNAs (DEMs) were screened out. Pathway analysis showed that upregulated genes were mainly enriched in the “B cell receptor signaling pathway”, “Cell cycle” and “NF-kappa B signaling pathway”, whereas downregulated genes were mainly enriched in the “Ribosome”, “FoxO signaling pathway” and “p53 signaling pathway”. We subsequently constructed a protein–protein interaction network of DEGs consisting of 277 genes and 563 interactions. In addition, 32 genes with high degrees in the network were identified as hub genes in MM, e.g. HDAC2, RBBP4, CREB1, and RB1. Additionally, we constructed a microRNA–mRNA regulatory network depicting interactions between DEMs and their targets, including the miR-135b–GADD45A and miR-148a–USPL1 pairs. In conclusion, the results of this data mining and integration help reveal the molecular basis of MM pathogenesis as well as potential biomarkers and therapeutic targets for MM diagnosis and treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma (MM), a common hematological malignancy, is characterized by clonal proliferation of malignant plasma cells in the bone marrow and a series of clinical manifestations, such as hypercalcemia, renal failure, anemia and bone lesions [1]. It usually evolves from its precancerous stage termed as monoclonal gammopathy of undetermined significance (MGUS), which is present in 2% of individuals over the age of 50 [2]. Traditionally, patients with MM had a poor prognosis, with 5-year overall survival less than 50%. Currently, the introduction of proteasome inhibitors and immunomodulatory drugs have greatly improved the clinical outcome of MM patients, but MM still remains an incurable disease.
Recent advances in molecular cytogenetics studies have contributed to a significant progress in understanding the occurrence and development of MM. Nearly fifty percent of MM cases are identified as hyperdiploid (HD-MM), which is characterized by trisomy of chromosomes 3, 5, 7, 9, 11, 15, 19 and 21. The remaining MM are referred as nHD-MM, harboring chromosomal translocations involving immunoglobulin heavy chains (IGH) genes, mainly t(4;14), t(6;14), t(11;14), t(14;16) and t(14;20) [3]. Additionally, a variety of somatic mutations, including NRAS, KRAS, BRAF and TP53 mutations, have been observed in MM [3–5]. What’s more, specific microRNAs (miRNAs) were shown to be deregulated in distinct subgroups of MM, mainly in association with IGH translocations or allelic imbalances [6, 7], suggesting that miRNAs exert a critical role in MM.
Microarray technology has been widely used for the investigation of general genetic aberrations involved in MM. However, there are few studies integrating these microarray datasets to identify keys genes and elucidate miRNA–mRNA regulatory networks in MM. Herein, we analyzed four sizeable and representative microarray profiles, including two gene expression profiles and two miRNA expression profiles, to obtain differentially expressed genes (DEGs) and differentially expressed miRNAs (DEMs) between MM and normal plasma cells. Additionally, functional enrichment analysis and protein–protein interaction (PPI) analysis were performed for DEGs, combined with miRNA–mRNA interaction analysis, to uncover potential mechanisms of MM occurrence and development.
Materials and methods
Microarray data
Two gene expression profiles (GSE16558 and GSE47552) and two miRNA expression profiles (GSE16558 and GSE17498) were downloaded from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo). GEO database is a functional genomics data repository, storing microarray and sequencing data, which is accessible to the public. We excluded MGUS, smoldering MM and plasma cell leukemia (PCL) patients in these datasets, which means only MM patients and healthy controls were preserved in this study. The two gene expression profiles included 101 MM samples and 10 normal plasma cell samples in total, whereas the two miRNA expression profiles included 98 MM samples and 8 normal plasma cell samples in total.
Data processing
GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) is an online program that enables investigators to perform comparison on two or more groups of samples in a GEO dataset to identify differentially expressed genes across experimental conditions. GEO2R analyzes original submitter-supplied processed microarray data using the GEOquery and limma R packages from the Bioconductor project. In present study, GEO2R was applied to obtain DEGs and DEMs in MM compared with normal plasma cells. The false positive result of microarray was then corrected by adjusted P value (adj. P value) using Benjamini–Hochberg method. The “adj. P value <0.05” and “|logFC| > 1” were set as the cut-off criterion.
Gene ontology (GO) and pathway analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/) is a web resource that offers functional interpretation of plenty of genes derived from genomic researches. In present study, DAVID database was used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The ontology contains three hierarchies: biological process, cellular component and molecular function. Pathway analysis is a functional analysis that maps genes to KEGG pathways. Herein, we performed GO analysis and KEGG pathway analysis using only overlapped differentially expressed genes in the two independent datasets (GSE16558 and GSE47552). The P value denoted the significance of the GO and pathway term enrichment in the DEGs. “P value <0.05” was set as the cut-off criterion.
Establishment of PPI network and clusters selection
In present study, protein–protein interactions (PPI) network of DEGs was constructed using Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, http://string.embl.de/). STRING is a biological database of known and predicted protein–protein interactions (PPI), which can provide system-wide view of cellular processes. The data is integrated and weighted, and a confidence score is calculated for all protein–protein interactions. Herein, we established the PPI network using only overlapped differentially expressed genes in the two independent datasets (GSE16558 and GSE47552). “Confidence score ≥0.7” was set as the cut-off criterion. To find clusters of genes in PPI network, Molecular Complex Detection (MCODE) was applied subsequently. “Degree cutoff = 2”, “node score cutoff = 0.2”, “k-core = 2” and “max. depth = 100” were set as the cut-off criterion.
Prediction of miRNA targets
The target genes of differentially expressed miRNAs were predicted using Targetscan (http://www.targetscan.org/), which is an online program that predicts targets of miRNAs by seeking the specific sequence complementary to the seed region of each miRNA. Predicted targets are ranked according to the predicted efficacy of targeting as calculated using cumulative weighted context++ scores of the sites. In present study, the top 100 genes were selected as target genes of each miRNA. Subsequently, miRNA–mRNA regulatory network depicting interactions between miRNAs and their potential targets in MM was visualized using Cytoscape.
Results
Identification of DEGs and DEMs
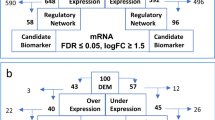
GEO2R analysis showed that a total of 866 and 873 DEGs were obtained in GSE16558 and GSE47552 datasets, respectively. Moreover, 601 DEGs were identified in both datasets. Among them, 596 genes showed consistent expression trends in both two datasets, including 514 upregulated genes and 82 downregulated genes in MM compared with normal plasma cells (Fig. 1a–c).

Identification of differentially expressed genes (a), upregulated genes (b) and downregulated genes (c) in mRNA expression profiling datasets GSE16558 and GSE47552. Identification of differentially expressed microRNAs (d) in microRNA expression profiling datasets GSE17498 and GSE16558
Additionally, our results showed that a total of 25 and 17 DEMs were screened out from GSE17498 and GSE16558 datasets, respectively. In GSE17498 dataset, 18 miRNAs were upregulated, and 7 miRNAs were downregulated in MM. In GSE16558 dataset, 17 miRNAs were upregulated in MM, but no downregulated miRNAs were observed. Among them, miR-155 and miR-135b were the most significantly upregulated miRNAs in GSE17498 and GSE16558 datasets, respectively, whereas miR-148a was the most significantly downregulated miRNA in GSE17498 dataset. What’s more, three miRNAs, miR-155, miR-203 and miR-486, were deregulated in both two datasets (Fig. 1d).
Functional enrichment analysis
GO analysis showed that the most significantly enriched GO terms corresponded to upregulated genes were “protein transport” (ontology: BP), “cytosol” (ontology: CC) and “protein binding” (ontology: MF), whereas the most significantly enriched GO terms corresponded to downregulated genes were “SRP-dependent cotranslational protein targeting to membrane” (ontology: BP), “extracellular exosome” (ontology: CC) and “structural constituent of ribosome” (ontology: MF) (Fig. 2).

Enriched GO terms for upregulated genes: a biological process (BP); b cellular component (CC); c molecular function (MF). Enriched GO terms for downregulated genes: d biological process (BP); e cellular component (CC); f molecular function (MF)
Further more, KEGG pathway analysis showed that the upregulated genes were enriched in 14 pathways, such as “B cell receptor signaling pathway”, “Cell cycle” and “NF-kappa B signaling pathway”, whereas the downregulated genes were enriched in 6 pathways, such as “Ribosome”, “FoxO signaling pathway” and “p53 signaling pathway” (Fig. 3).

Enriched pathways corresponding to upregulated genes (a). Enriched pathways corresponding to downregulated genes (b)
Establishment of PPI network and identification of hub genes
The PPI network of DEGs was constructed by STRING, which was composed of 277 nodes and 563 edges, including 234 upregulated genes and 43 downregulated genes (Fig. 4). Moreover, 32 genes in the PPI network were identified as hub genes in MM, e.g. HDAC2, RBBP4, CREB1 and RB1 genes, when “Degrees ≥10” was set as the cut-off criterion.

Protein–protein interaction network of differentially expressed genes
Subsequently, 16 clusters were selected from PPI network using MCODE, and it was shown that the most significant cluster consisted of 12 nodes and 61 edges. Besides, MCODE analysis showed that each cluster contained one “seed” gene, for example, EIF5B, KNTC1, IFI6, SOS1 and UTP6 genes were identified as the “seed” genes in their own clusters, respectively (Fig. 5).

Five significant modules selected from protein–protein interaction network
Construction of microRNA-target regulatory network
The target genes of 39 DEMs were predicted by Targetscan. By comparing target genes with DEGs, we found that 31 DEMs, including 25 upregulated and 6 downregulated miRNAs, presented an expression trend opposite to that of their predicted targets in MM. In our study, miR-135b was the most significantly upregulated miRNAs in GSE16558 datasets and was predicted to target GADD45A and RNF7 genes. MiR-148a was the most significantly downregulated miRNA in GSE17498 dataset and was predicted to target 7 upregulated genes, including ACADM, NIN, AGL, ERLIN1, MICU2, USPL1 and ESM1 genes. MiR-363 was downregulated and was predicted to target 9 upregulated genes, including KLHL14, GPR180, ARMC1, UBXN4, SNAPC1, IDH1, USP28, AARS and KIAA1109 genes. Moreover, we noticed that several deregulated genes were predicted to be the common targets of different miRNAs. For instance, CCL3 gene was predicted as the common target of miR-196a and miR-484. ATF2 gene was predicted as the common target of miR-29c and miR-451 (Fig. 6).

MicroRNA–mRNA regulatory networks in multiple myeloma
Discussion
Multiple myeloma is a genetically complex and heterogeneous disease resulting from multiple genomic events that lead to tumor development and progression. Alongside the introduction of novel therapies, the outcome of MM has greatly improved. However, this improvement is not identical among all patients with MM, partly due to the genomic complexity of MM. Therefore, investigation of the molecular mechanisms of MM becomes urgent and essential. Recently, the rapid development and wide use of microarray technology have contributed a lot to uncover the general genetic alterations of MM. However, there is almost no study integrating these datasets together.
In present study, a total of 596 DEGs were identified from GSE16558 and GSE47552 datasets, consisting of 514 upregulated genes and 82 downregulated genes. The upregulated genes were enriched in 14 pathways, such as “B cell receptor signaling pathway”, “Cell cycle” and “NF-kappa B signaling pathway”, whereas downregulated genes were enriched in 6 pathways, such as “Ribosome”, “FoxO signaling pathway” and “p53 signaling pathway”. Among the 596 DEGs, 32 genes were identified as hub genes in MM, e.g. HDAC2, RBBP4, CREB1 and RB1 genes, due to their high degrees in PPI network. Additionally, 16 clusters were obtained from the PPI network using MCODE.
Apart from the central role of genetic aberrations, epigenetic abnormalities have been shown to be important players in MM. Recently, many studies have focused on the roles of DNA methylation, histone modifications and noncoding RNAs in MM [8–10]. Herein, we investigated previously published GEO datasets of MM and then identified several critical deregulated genes associated with epigenetic modifications in MM via IPI network, including HDAC2, RBBP4 and KAT2B genes.
HDAC2 is a member of histone deacetylases (HDACs) family, which can catalyze core histones deacetylation, resulting in chromatin condensation and transcription inhibition [11, 12]. Increasing evidence has demonstrated aberrant expression of HDAC2 in neoplastic disease, leading to aberrant gene expression and cellular signaling that promote cell cycle progression, cell growth and resistance to apoptosis. Specifically, combination treatment with HDAC inhibitors and proteasome inhibitors or immunomodulatory drugs has shown remarkable anti-MM activity in both preclinical and clinical studies [13]. HDAC2 epigenetically silences the expression of pro-apoptotic protein NOXA in pancreatic cancer cells [14] and APAF1 in colon cancer cells [15]. Additionally, HDAC2 regulates the transcriptional activities of p53 through modulating p53-DNA binding activity [16]. Moreover, HDAC2 and HDAC1 cooperate in regulating the expression of DNA repair proteins BRCA1, CHK1 and RAD51 in acute myeloid leukemia cells [17]. RBBP4 is a component of several chromatin modifying protein complexes with varying effects on gene expression. RBBP4 contributes to repression of gene transcription as a key member of nucleosome remodeling and deacetylation (NuRD) complex, polycomb repressor complex 2 (PRC2) and chromatin assembly factor 1 (CAF1) complex. NuRD complex can promote histone deacetylation and nucleosome remodeling [18]. PRC2 complex can methylate Lys-9 and Lys-27 of histone H3 [19]. CAF1 complex is required for chromatin assembly following DNA replication and DNA repair [20, 21]. Therefore, we inferred that HDAC2 and RBBP4 could function as epigenetic modulators and subsequently affect the biological behavior and response to therapies in MM.
It has been shown that miRNAs induce RNA silencing by targeting 3′-UTR of mRNAs, and that abnormal expression of miRNAs contributes to occurrence of various cancer types, including MM [6, 9]. In this study, we identified 31 DEMs, including 25 upregulated and 6 downregulated miRNAs in MM. Among them, miR-155 and miR-135b were the most significantly upregulated miRNAs in GSE17498 and GSE16558 datasets, respectively, whereas miR-148a was the most significantly downregulated miRNA in GSE17498 dataset.
MiR-135b was shown to be overexpressed in various tumors, such as hepatocellular carcinoma [22], colorectal cancer [23, 24] and breast cancer [25]. In our study, GADD45A was predicted as the target of miR-135b, which was downregulated in MM according to GSE16558 and GSE47552 datasets. GADD45 family play an important role in cell cycle control, cell growth, apoptosis, maintenance of genomic stability, DNA repair and active DNA demethylation in response to environmental and physiological stress [26, 27]. GADD45A behaves as a tumor suppressor in many cancers, such as prostate cancer and gastric cardia adenocarcinoma [28, 29]. Downregulated expression of GADD45A is often associated with promoter methylation [28, 29] and elevated MYC [30]. Herein, we proposed that miR-135b might function as an oncogenic factor and suppress GADD45A expression by targeting its 3′-UTR in MM. It’s worth noting that the results of miR-155 expression level in MM remain controversial based on previous studies. MiR-155 was shown to be epigenetically silenced by DNA methylation in MM [31, 32], and ectopic expression of miR-155 significantly inhibited MM cell proliferation and colony formation [32]. However, Marta et al. suggested that miR-155 was expressed at higher levels in patients with either t(14;16) or t(14;20) translocations than other MM patients [7]. These conflicting results showed that the expression pattern of miR-155 may be associated with distinct molecular groups of MM. Maybe more work should be done in the future to illustrate the relationship between miR-155 expression and MM subtypes.
MiR-148a is implicated in various biological processes, including cellular differentiation and development. MiR-148a expression increases after pre-B cell activation and then promotes differentiation of activated B cells to plasma cells [33]. MiR-148a was shown to be downregulated in many cancers, including gastric [34, 35], colorectal [36] and pancreatic cancers [37]. In our study, miR-148a was predicted to target 7 genes in MM, and USPL1 was one of them, which was upregulated in MM according to GSE16558 and GSE47552 datasets. Posttranslational modification of proteins by covalent conjugation of small ubiquitin-like modifiers (SUMO), termed SUMOylation, is an essential cellular process [38–40]. USPL1 is a newly discovered SUMO-specific isopeptidase, which removes SUMO from targets and is involved in various nuclear functions ranging from maturation of snRNPs to telomere integrity [41–43]. Genetic variation in USPL1 was reported to contribute to risk for grade-3 breast cancer recently [44]. It was the first time for USPL1 to be linked to malignancy. In present study, USPL1 was predicted as one of the targets of miR-148a, which indicated that miR-148a might behave as a tumor suppressor and participate in posttranslational modification of proteins in MM by regulating USPL1 expression.
Additionally, we found that a single miRNA could target several genes in MM. MiR-363 was predicted to target 9 genes, including KLHL14, GPR180, ARMC1, UBXN4, SNAPC1, IDH1, USP28, AARS and KIAA1109 genes. MiR-148a was predicted to target 7 genes, including ACADM, NIN, AGL, ERLIN1, MICU2, USPL1 and ESM1 genes. Similarly, a single gene could be the common target of several miRNAs in MM. CCL3 was predicted as the target of miR-196a and miR-484, and ATF2 was predicted as the target of miR-29c and miR-451.
In conclusion, our study was intended to identify key genes in MM and construct regulatory networks between miRNAs and mRNAs through bioinformatics analysis. HDAC2 and RBBP4 were identified as key epigenetic-related genes in MM according to PPI network, which were associated with various epigenetic modifications ranging from histone modifications to chromatin assembly. Additionally, deregulated miRNAs in MM, such as miR-135b and miR-148a, might exert their biological functions through targeting 3′-UTR of mRNAs. Our study indicated that data mining and integration was an effective method to investigate the molecular pathogenesis of MM. Moreover, our results could provide novel diagnostic biomarkers and therapeutic targets for MM patients.
References
Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046–60. doi:10.1056/NEJMra1011442.
Landgren O, Kristinsson SY, Goldin LR, Caporaso NE, Blimark C, Mellqvist UH, et al. Risk of plasma cell and lymphoproliferative disorders among 14621 first-degree relatives of 4458 patients with monoclonal gammopathy of undetermined significance in Sweden. Blood. 2009;114(4):791–5. doi:10.1182/blood-2008-12-191676.
Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6:6997. doi:10.1038/ncomms7997.
Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91–101. doi:10.1016/j.ccr.2013.12.015.
Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. doi:10.1038/ncomms3997.
Rio-Machin A, Ferreira BI, Henry T, Gomez-Lopez G, Agirre X, Alvarez S, et al. Downregulation of specific miRNAs in hyperdiploid multiple myeloma mimics the oncogenic effect of IgH translocations occurring in the non-hyperdiploid subtype. Leukemia. 2013;27(4):925–31. doi:10.1038/leu.2012.302.
Lionetti M, Biasiolo M, Agnelli L, Todoerti K, Mosca L, Fabris S, et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood. 2009;114(25):e20–6. doi:10.1182/blood-2009-08-237495.
Sharma A, Heuck CJ, Fazzari MJ, Mehta J, Singhal S, Greally JM, et al. DNA methylation alterations in multiple myeloma as a model for epigenetic changes in cancer. Wiley Interdiscip Rev Syst Biol Med. 2010;2(6):654–69. doi:10.1002/wsbm.89.
Dimopoulos K, Gimsing P, Gronbaek K. Aberrant microRNA expression in multiple myeloma. Eur J Haematol. 2013;91(2):95–105. doi:10.1111/ejh.12124.
Mithraprabhu S, Kalff A, Chow A, Khong T, Spencer A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics. 2014;9(11):1511–20. doi:10.4161/15592294.2014.983367.
Brandl A, Heinzel T, Kramer OH. Histone deacetylases: salesmen and customers in the post-translational modification market. Biol Cell. 2009;101(4):193–205. doi:10.1042/BC20080158.
Kramer OH. HDAC2: a critical factor in health and disease. Trends Pharmacol Sci. 2009;30(12):647–55. doi:10.1016/j.tips.2009.09.007.
Harada T, Hideshima T, Anderson KC. Histone deacetylase inhibitors in multiple myeloma: from bench to bedside. Int J Hematol. 2016;104(3):300–9. doi:10.1007/s12185-016-2008-0.
Fritsche P, Seidler B, Schuler S, Schnieke A, Gottlicher M, Schmid RM, et al. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut. 2009;58(10):1399–409. doi:10.1136/gut.2009.180711.
Hanigan CL, Van Engeland M, De Bruine AP, Wouters KA, Weijenberg MP, Eshleman JR, et al. An inactivating mutation in HDAC2 leads to dysregulation of apoptosis mediated by APAF1. Gastroenterology. 2008;135(5):1654–64. doi:10.1053/j.gastro.2008.07.078.
Wagner T, Brand P, Heinzel T, Kramer OH. Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim Biophys Acta. 2014;1846(2):524–38. doi:10.1016/j.bbcan.2014.07.010.
Zhao J, Xie C, Edwards H, Wang G, Taub JW, Ge Y. Histone deacetylases 1 and 2 cooperate in regulating BRCA1, CHK1, and RAD51 expression in acute myeloid leukemia cells. Oncotarget. 2016;. doi:10.18632/oncotarget.14062.
Todd MA, Picketts DJ. PHF6 interacts with the nucleosome remodeling and deacetylation (NuRD) complex. J Proteome Res. 2012;11(8):4326–37. doi:10.1021/pr3004369.
Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16(22):2893–905. doi:10.1101/gad.1035902.
Furuyama T, Dalal Y, Henikoff S. Chaperone-mediated assembly of centromeric chromatin in vitro. Proc Natl Acad Sci USA. 2006;103(16):6172–7. doi:10.1073/pnas.0601686103.
Zhang W, Tyl M, Ward R, Sobott F, Maman J, Murthy AS, et al. Structural plasticity of histones H3–H4 facilitates their allosteric exchange between RbAp48 and ASF1. Nat Struct Mol Biol. 2013;20(1):29–35. doi:10.1038/nsmb.2446.
Li Y, Xu D, Bao C, Zhang Y, Chen D, Zhao F, et al. MicroRNA-135b, a HSF1 target, promotes tumor invasion and metastasis by regulating RECK and EVI5 in hepatocellular carcinoma. Oncotarget. 2015;6(4):2421–33. doi:10.18632/oncotarget.2965.
He Y, Wang J, Wang J, Yung VY, Hsu E, Li A, et al. MicroRNA-135b regulates apoptosis and chemoresistance in colorectal cancer by targeting large tumor suppressor kinase 2. Am J Cancer Res. 2015;5(4):1382–95.
Wu W, Wang Z, Yang P, Yang J, Liang J, Chen Y, et al. MicroRNA-135b regulates metastasis suppressor 1 expression and promotes migration and invasion in colorectal cancer. Mol Cell Biochem. 2014;388(1–2):249–59. doi:10.1007/s11010-013-1916-z.
Hua K, Jin J, Zhao J, Song J, Song H, Li D, et al. miR-135b, upregulated in breast cancer, promotes cell growth and disrupts the cell cycle by regulating LATS2. Int J Oncol. 2016;48(5):1997–2006. doi:10.3892/ijo.2016.3405.
Hoffman B, Liebermann DA. Gadd45 modulation of intrinsic and extrinsic stress responses in myeloid cells. J Cell Physiol. 2009;218(1):26–31. doi:10.1002/jcp.21582.
Liebermann DA, Tront JS, Sha X, Mukherjee K, Mohamed-Hadley A, Hoffman B. Gadd45 stress sensors in malignancy and leukemia. Crit Rev Oncogenesis. 2011;16(1–2):129–40.
Guo W, Dong Z, Guo Y, Chen Z, Kuang G, Yang Z. Methylation-mediated repression of GADD45A and GADD45G expression in gastric cardia adenocarcinoma. Int J Cancer. 2013;133(9):2043–53. doi:10.1002/ijc.28223.
Ramachandran K, Gopisetty G, Gordian E, Navarro L, Hader C, Reis IM, et al. Methylation-mediated repression of GADD45alpha in prostate cancer and its role as a potential therapeutic target. Cancer Res. 2009;69(4):1527–35. doi:10.1158/0008-5472.CAN-08-3609.
Amente S, Zhang J, Lavadera ML, Lania L, Avvedimento EV, Majello B. Myc and PI3 K/AKT signaling cooperatively repress FOXO3a-dependent PUMA and GADD45a gene expression. Nucleic Acids Res. 2011;39(22):9498–507. doi:10.1093/nar/gkr638.
Krzeminski P, Sarasquete ME, Misiewicz-Krzeminska I, Corral R, Corchete LA, Martin AA, et al. Insights into epigenetic regulation of microRNA-155 expression in multiple myeloma. Biochim Biophys Acta. 2015;1849(3):353–66. doi:10.1016/j.bbagrm.2014.12.002.
Bi C, Chung TH, Huang G, Zhou J, Yan J, Ahmann GJ, et al. Genome-wide pharmacologic unmasking identifies tumor suppressive microRNAs in multiple myeloma. Oncotarget. 2015;6(28):26508–18. doi:10.18632/oncotarget.4769.
Porstner M, Winkelmann R, Daum P, Schmid J, Pracht K, Corte-Real J, et al. miR-148a promotes plasma cell differentiation and targets the germinal center transcription factors Mitf and Bach2. Eur J Immunol. 2015;45(4):1206–15. doi:10.1002/eji.201444637.
Chen Z, Saad R, Jia P, Peng D, Zhu S, Washington MK, et al. Gastric adenocarcinoma has a unique microRNA signature not present in esophageal adenocarcinoma. Cancer. 2013;119(11):1985–93. doi:10.1002/cncr.28002.
Zheng G, Xiong Y, Xu W, Wang Y, Chen F, Wang Z, et al. A two-microRNA signature as a potential biomarker for early gastric cancer. Oncol Lett. 2014;7(3):679–84. doi:10.3892/ol.2014.1797.
Takahashi M, Cuatrecasas M, Balaguer F, Hur K, Toiyama Y, Castells A, et al. The clinical significance of MiR-148a as a predictive biomarker in patients with advanced colorectal cancer. PLoS One. 2012;7(10):e46684. doi:10.1371/journal.pone.0046684.
Hanoun N, Delpu Y, Suriawinata AA, Bournet B, Bureau C, Selves J, et al. The silencing of microRNA 148a production by DNA hypermethylation is an early event in pancreatic carcinogenesis. Clin Chem. 2010;56(7):1107–18. doi:10.1373/clinchem.2010.144709.
Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8(12):947–56. doi:10.1038/nrm2293.
Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11(12):861–71. doi:10.1038/nrm3011.
Johnson ES. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355–82. doi:10.1146/annurev.biochem.73.011303.074118.
Hay RT. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 2007;17(8):370–6. doi:10.1016/j.tcb.2007.08.002.
Bawa-Khalfe T, Yeh ET. SUMO losing balance: SUMO proteases disrupt SUMO homeostasis to facilitate cancer development and progression. Genes Cancer. 2010;1(7):748–52. doi:10.1177/1947601910382555.
Schulz S, Chachami G, Kozaczkiewicz L, Winter U, Stankovic-Valentin N, Haas P, et al. Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Rep. 2012;13(10):930–8. doi:10.1038/embor.2012.125.
Bermejo JL, Kabisch M, Dunnebier T, Schnaidt S, Melchior F, Fischer HP, et al. Exploring the association between genetic variation in the SUMO isopeptidase gene USPL1 and breast cancer through integration of data from the population-based GENICA study and external genetic databases. Int J Cancer. 2013;133(2):362–72. doi:10.1002/ijc.28040.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
About this article

Cite this article
Gao, H., Wang, H. & Yang, W. Identification of key genes and construction of microRNA–mRNA regulatory networks in multiple myeloma by integrated multiple GEO datasets using bioinformatics analysis. Int J Hematol 106, 99–107 (2017). https://doi.org/10.1007/s12185-017-2216-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2216-2