
Abstract
Polycomb repressive complex (PRC) is a critical regulator of normal tissue homeostasis as well as tumorigenesis. EZH2, an enzymatic subunit of PRC2, is a histone H3K27 methyltransferase that functions in the regulation of gene silencing. EZH2 overexpression was first identified in prostate and breast cancers and is associated with poor clinical outcome. Subsequently, gain- and loss-of-function mutations of EZH2 have been identified in various tumors, including hematological malignancies, implicating EZH2 as either an oncogene or a tumor suppressor gene, depending on the cancer type. Molecular mechanisms underlying the multifaceted function of EZH2 have been analyzed extensively. However, because EZH2 dysregulation is functionally integrated with multiple other epigenetic events in a context-dependent manner, the precise manner in which EZH2 dysregulation impacts the pathogenesis of hematological malignancies remains to be clarified. In this perspective, we describe recent findings in pathogenic role of EZH2 in hematological malignancies, which may provide insights into the treatment of with cancers with EZH2 dysregulation and the development of novel therapies targeting epigenetic regulators.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polycomb group (PcG) proteins were initially identified in Drosophila as regulators of body segmentation that function by the repression of homeotic genes and were subsequently identified in mammals [1]. PcG proteins function in the maintenance of gene silencing via histone modifications. This repressive function of PcG in transcription is biologically counteracted by the activating function of Trithorax-group (TrxG) complexes (e.g. H3K4 methyltransferase MLL1) and maintains proper regulation of gene expression in development and adult tissues homeostasis [2]. The function of PcG proteins is often dysregulated in cancer, mainly through altered expression levels and somatic gene mutations. Dysregulation of EZH2 of the Polycomb Repressive Complex 2 (PRC2) is strongly oncogenic and has been extensively analyzed. In this review, we focus on recent findings on the pathological role of EZH2 in hematological malignancies and the therapeutic impact of targeting EZH2 enzymatic activity in cancers.
Role of PRC2 in normal hematopoiesis
In mammals, there are two major complexes of PcG complexes: PRC 1 and PRC2 (Fig. 1a). PRC2 contains three core subunits; SUZ12, EED, and either of the two histone H3K27 methyltransferases, EZH1 or EZH2, which catalyze mono-, di- and tri-methylation of histone H3 at lysine 27 (H3K27me1/me2/me3). Following trimethylation of H3K27 by PRC2, canonical PRC1 is subsequently recruited via binding of CBX, a subunit of PRC1, to H3 K27me3. Canonical PRC1 contains four core subunits: PCGF, CBX, PHC, and one of the two histone H2AK119 mono-ubiquitylases, RING1A or RING1B, which exhibit E3 ubiquitin ligase activity on histone H2A at lysine 119 (H2AK119ub1). H2AK119ub1 modification has a role in the consolidation of repression by inhibiting transcriptional elongation and promoting chromatin compaction [3]. In addition to canonical PRC1, there are at least four PRC1 variants that also show monoubiquitylation activity at H2AK119 and which can function independently of PRC2 [4].

EZH2Y641 mutant re-distributes H3K27me3, resulting in both repression and activation of PRC2-target genes. a Schematic representation of PRC1 and PRC2. b Ezh2Y641 mutant enhances levels of H3K27me3 at promoters and gene body regions, leading to silencing expression of genes involved in GC B-cell differentiation and cell cycle checkpoint. After modifications of the mono-methylation of H3K27 by wild-type Ezh2-PRC2, EZH2Y641-PRC2 modifies H3K27me2/me3 due to its higher di- and tri-methylation activity. c Ezh2Y641 mutant de-represses expression of many Ezh2-PRC2 targets in normal B-cells (e.g. Hoxc4, Hoxc9 and Meis1) and represses many genes lacking H3K27me3 in normal B cells. However, it is unknown how mutant Ezh2Y641 impairs wild-type Ezh2-PRC2 function at certain regions. In the figure, Y641 indicates Ezh2Y641 mutant
PRC2 is a critical regulator of normal hematopoiesis [5, 6]. Loss of EED function profoundly compromises adult, but not fetal, hematopoiesis due to impaired repopulation and differentiation of hematopoietic stem cells (HSCs) in mice [7]. Ezh1 prevents premature senescence of HSCs via silencing expression of Cdkn2a, a major target of the PcG complexes [8]. In contrast, Ezh2 is dispensable for self-renewal of HSCs due to the compensatory function of Ezh1. Ezh1-containing PRC2 (Ezh1-PRC2) co-regulates a large number of target genes with Ezh2-containing PRC2 (Ezh2-PRC2) and, notably, is redistributed to a significant portion of Ezh2-specific targets on the loss of Ezh2 [7, 9, 10]. On overexpression, Ezh2 efficiently prevents exhaustion of the long-term repopulating potential of HSCs during repeated serial transplantation [11]. Hematopoietic cell-specific overexpression of Ezh2 in mice also augmented HSC function, resulting in the development of myeloproliferative neoplasms (MPNs) [12]. These findings indicate the distinct, but equally important, roles of Ezh1 and Ezh2 in the regulation of hematopoiesis.
Overexpression of EZH2 in solid tumors: is EZH2 a functional oncogene?
Overexpression of wild-type EZH2 was initially identified in prostate cancer and is associated with disease progression and poor prognosis [13]. Similar effects have also been confirmed in a variety of cancers, including breast cancer, bladder cancer, endometrial cancer, and melanoma [14]. In patients with these cancers, levels of EZH2 protein are strongly associated with capacities of tumor proliferation [14]. EZH2 transcription is directly activated by E2F family transcription factors, major targets of the retinoblastoma protein (RB) [15], and by ETS transcription factor ERG or TMPRSS2-ERG fusion protein in prostate cancer [16]. Transcription of EZH2 is also directly or indirectly activated by c-MYC, a potent oncogene [17, 18]. In turn, EZH2 regulates p53 and p16Ink4a-RB tumor suppressor pathways thorough repressing expression of CDKN2A, suggesting that EZH2 acts as an oncogene in these contexts.
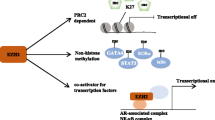
Many studies have focused on the PRC2-mediated repression of target genes, such as tumor suppressor and differentiation-related genes, as the oncogenic function of EZH2. However, recent studies have also demonstrated a role for EZH2 independent of the methylation of H3K27. Several studies have shown that EZH2 plays a transcriptional co-activator in concert with NF-κB or androgen receptor (AR) in certain cancers [19, 20]. In castration-resistant prostate cancer cells, phosphorylation of EZH2 at Ser21, mediated directly or indirectly by the PI3K-Akt pathway, can switch its function from a Polycomb repressor to a transcriptional coactivator of AR. In this setting, EZH2 still requires its methyltransferase activity, but targets substrates other than H3K27, potentially non-histone proteins. Similarly, while SWI/SNF-mutant cancer cells are primarily dependent on the non-catalytic role of EZH2, they also depend in part on its enzymatic activity [21]. Given these findings, it remains unclear whether elevated expression of EZH2 actually contributes to tumorigenesis through enhanced enzymatic activity of EZH2-PRC2.
In contrast, it has recently been reported that the PRC2 components, SUZ12 and EED, are frequently deleted or mutated in malignant peripheral nerve sheath tumors (MPNST) [22]. PRC2 loss usually co-occurs with NF1 deletion and potentiates the effects of NF1 deletion by amplifying Ras-driven transcription through enhancement of H3K27 acetylation at transcriptional regulatory regions following the loss of H3K27me3. It has also been reported that Ezh2 is dispensable in genetically engineered mouse models of breast cancers and that genetic disruption of EZH2 in a breast cancer cell line promotes tumorigenesis [23]. In breast cancer, low levels of EZH2, resulting from genetic loss of EZH2 and mutations in PRC2 genes, are associated with poor prognosis. In pediatric high-grade glioma (HGG), histone H3 is frequently mutated. H3K27 M mutation is identified up to in 78% of diffuse intrinsic pontine gliomas and also at high frequencies in non-brainstem gliomas. H3K27 M mutation likely stabilizes the binding of PRC2 to H3K27 M, thereby preventing deposition of methyl marks on other H3. Total H3K27me2/3 levels are thus profoundly reduced in cells expressing the K27M mutant allele, resulting in mis-regulation of PRC2 target genes [24]. All these findings indicate the tumor suppressive role of EZH2-PRC2 in a subset of solid tumors.
Oncogenic Function of EZH2Y641 mutation in B cell malignancies
Comprehensive genome sequencing analyses identified gain-of-function mutations of EZH2Y641 (Y641E, Y641F, Y641 N, Y641S, Y641C, and Y641H) in 30% of germinal center (GC)-like diffuse large B-cell lymphoma (DLBCL) and 10% of follicular B-cell lymphoma [25]. EZH2Y641 mutations in the catalytic SET domain have higher di- and tri-methylation activity than wild-type EZH2, but significantly lower mono-methylation activity, due to its impaired recognition of unmodified H3K27 [26]. These lymphoma cells always harbor a single mutant allele and show higher H3K27me3 levels than lymphoma cells with wild-type EZH2. Thus, wild-type and mutant alleles collaborate to efficiently tri-methylate H3K27, promoting the development of lymphoma [27]. Another EZH2 activating mutation of alanine 677 (A677G), which was identified in B-cell lymphoma, also shows increased levels of H3K27me3 [28].
In normal B-lymphopoiesis, Ezh2 is important for VDJ recombination in pre-B cells, but its expression declines and is undetectable in mature B-cells [29]. When B-cells enter the GC reaction, Ezh2 expression is once again activated, and indeed, Ezh2 has been shown to be required for GC formation by conditional deletion of Ezh2 in GC B cells in mouse [30]. While expression of EZH2 Y641N driven by a collagen promoter induced GC hyperplasia with increased levels of H3K27me3, it did not induce GC-like lymphoma in mice [30]. In contrast, expression of EZH2 Y641F driven by the endogenous Ezh2 locus induced the formation of lymphoma with a median survival of one year [31]. EZH2Y641 mutants may cooperate with co-existing genetic mutations in lymphomagenesis. Indeed, co-expression of BCL2 oncogene or loss of Trp53 tumor suppressor significantly accelerated the formation of EZH2 Y641-induced lymphoma in both mouse models [30, 31]. In addition, both shRNA-mediated knockdown of Ezh2 and chemical inhibition of Ezh2 enzymatic activity significantly impaired lymphoma cell growth in vivo [31]. These findings clearly indicate that Ezh2-PRC2 function is required for both initiation and maintenance of lymphoma.
Detailed molecular functions of EZH2Y641 mutants have recently been described by utilizing Ezh2 Y641 knock-in mice (Table 1). Corresponding to the physiological role for Ezh2 in the maintenance of GC B cells, EZH2Y641 mutants preferentially repressed expression of genes involved in the exit from GC and terminal differentiation (e.g. Prdm1 and Irf4) and cell cycle checkpoint genes (e.g. Cdkn1a and Cdkn1b) via PRC2-mediated H3K27me3 at their promoter regions [30]. Recently, it has also been shown that wild-type as well as Ezh2Y641 mutants cooperate with Bcl6 to mediate combinatorial tethering of a non-canonical PRC1-Bcor-Cbx8 complex to target genes, resulting in the consolidation of silencing of Prdm1 and Irf4 expression through additional H2K119ub1 modification [32]. Thus, Ezh2Y641F mutant significantly increases levels of H3K27me3 at regions which are directly repressed by Ezh2-PRC2 in normal B cells (Fig. 1b). Of importance, however, Ezh2Y641F mutant appeared to represses many genes, which lack H3K27me3 but marked with H3K27ac in normal B cells, and paradoxically induce expression of genes directly repressed by Ezh2-PRC2 with H3K27me3 in normal B cells such as Hoxc4, Hoxc9 and Meis1, which may contribute to formation of tumor (Fig. 1c) [31]. Genes repressed by Ezh2Y641F mutants show increased levels of H3K27me3 in the gene body regions, consistent with a previous report showing that H3K27me3 modifications in the gene body are strongly repressive [33]. Although it remains unknown how Ezh2Y641 mutants erase or add H3K27me3 modifications at certain regions, EZH2Y641 mutants induce a vast reorganization of chromatin structure and reprogram the transcriptional profiles.
Oncogenic function of EZH2 in de novo AML
In contrast to B-cell lymphoma, no activating mutations are found in EZH2 in de novo AML patients [34]. Loss-of-function mutations of EZH2 are also rare (about 1%) in de novo AML patients [34], but are common in MDS, where EZH2 mutations are again not associated with the progression to AML [35, 36]. Ezh2-deficient hematopoietic cells develop MDS and MDS/MPN-like diseases in mice, but not AML even in serially transplantation [9, 37], implying an oncogenic property of EZH2 in the pathogenesis of AML. In fact, deletion of Ezh2 resulted in significantly reduced leukemia-initiating cells and enhanced differentiation of leukemic cells in a mouse model of MLL-AF9 induced AML. In this AML model, Ezh2 was shown to repress developmental and differentiation regulators (e.g. Egr-1) [38, 39]. However, AML cells still retained leukemia-initiating cells even in the absence of Ezh2 (Table 1). Correspondingly, deletion of Eed resulted in complete loss of PRC2 function and abolished leukemogenecity of MLL-AF9 induced AML cells [38]. These findings indicate that the presence of Ezh2 is not strictly required for MLL-AF9 induced AML conceivably due to compensatory function of Ezh1; however, Ezh2-PRC2 contributes to the propagation of AML, at lease in part, due to silencing of PRC2 target genes to impede the spontaneous differentiation of leukemic stem cells.
Tumor suppressive function of EZH2 in MDS, MPN and T-ALL
Although over-expression and gain-of-function mutations of EZH2 play oncogenic roles in the development of many cancers, it has also become evident that EZH2 functions as a tumor suppressor in a number of hematological malignancies [40]. Deletions and missense and frameshift mutations in EZH2 that abrogate its methyltransferase activity are frequently observed in MDS (3–7%), MPN (3–13%) and MDS/MPN overlap disorders (8–13%), leading to reduction in H3K27me3 levels [41, 42]. In contrast to B-lymphoma cells carrying heterozygous mutation of EZH2 Y641, loss-of-function mutations were found both mono-and bi-allelically in myeloid malignancies. Indeed, MDS patients with mutations of EZH2 show significantly poor outcome compared to patients without mutations, and the survival of patients with homozygous mutations is relatively shorter than for those with heterozygous mutations [41]. Deletion of the long arm of chromosome 7 (7q-), a characteristic cytogenetic anomaly frequently observed in MDS, commonly involves EZH2 at 7q36 and is associated with very poor prognosis. A recent functional mapping study by utilizing 7q- MDS patient-derived iPS cells elegantly demonstrated that impaired hematopoiesis of 7q- MDS can be modestly rescued by exogenous EZH2, indicating a causative role of haploinsufficiency of EZH2 in combination with other 7q genes, such as LUC7L2, HIPK2 and ATP6V0E2, in defective hematopoiesis of 7q- MDS clones [43]. Primary myelofibrosis (PMF) is a subtype of MPN and is driven by JAK2 V617F activating mutation or other mutations that activate JAK2. EZH2 mutations have been shown to predict poor survival in patients with PMF regardless of the presence of JAK2 mutation [44], consistent with a tumor suppressive function of EZH2.
The molecular mechanisms underlying pathological roles of loss-of-function mutations of EZH2 have been described by utilizing Ezh2 knockout mice (Table 1). Hematopoietic cell-specific deletion of Ezh2 resulted in the development of various myeloid malignancies with long latencies, including MDS and MDS/MPN, while Ezh2-deficient cortical T-ALL was occasionally observed in the setting of serial transplantation [9]. TET2 mutations frequently coexist with EZH2 mutations in patients. When Ezh2-deficient cells (Ezh2 Δ/Δ) were combined with a hypomorphic mutant of Tet2 (Tet2 KD/KD), these Tet2 KD/KD Ezh2 Δ/Δ mutant mice exhibited significantly accelerated formation of MDS and MDS/MPN [45]. In addition, the loss of Ezh2 enhanced the initiation and progression of RUNX1 mutant-induced MDS, but attenuated the predisposition to leukemic transformation [37], consistent with an oncogenic effect of Ezh2 in MLL-AF9-induced AML. We and other groups have also demonstrated that Ezh2 loss significantly promotes the development of JAK2 V617F mutant-induced myelofibrosis (MF), at least in part, due to the enhancement of aberrant megakaryocytopoiesis, which is thought to be critical for the formation of fibrosis [46–48]. These results clearly indicate that EZH2 plays a tumor suppressive role in myelodysplastic and myeloproliferative disorders that originate from HSCs.
Loss-of-function mutations of EZH2 and SUZ12 have also been found in 25% of cortical T-cell acute lymphoblastic leukemia (T-ALL) [49]. Moreover, in mouse models, Ezh2-deficient hematopoietic cells are reported to induce T-ALL in addition to heterogeneous hematological malignancies [9, 50]. In a mouse model and human T-ALL cells, oncogenic NOTCH1 mutation specifically induces loss of H3K27me3 modification by antagonizing the function of PRC2, leading to the activation of NOTCH1 transcriptional program [49]. These findings indicate that PRC2 also functions as a tumor suppressor in T-ALL.
Mechanism of tumorigenesis driven by EZH2 dysfunction
Given that EZH2-PRC2 represses many target genes, EZH2 dysfunction is thought to de-repress expression of various potential oncogenes in cancer. Indeed, Ezh2 loss in JAK2V617F hematopoietic cells in mice promoted an “epigenetic switch” characterized by reduced levels of H3K27me3 followed by enhanced H3K27 acetylation (H3K27ac) at promoter regions (Fig. 2a), leading to the activation of 243 of 2,073 PRC2 target genes, including potential oncogenes such as Hmga2, Mlf1, and Pbx3 [46]. Activation of these potential oncogenes was also observed in Tet2 KD/KD Ezh2 Δ/Δ MDS cells. HMGA2 is significantly up-regulated in CD34+ cells in patients with PMF [47, 51], and overexpression of Hmga2 in JAK2V617F HSCs promoted the production of dysplastic megakaryocytes both in vitro and in vivo [47, 52]. These findings implicate HMGA2 in the pathogenesis of PMF. The epigenetic switch from H3K27me3 to H3K27ac is not limited to myeloid malignancies. As described above, deletion of NF1 RasGAP is involved in many cancers, including glioblastoma (GBM) and MPNST, and causes constitutive activation of Ras signaling. SUZ12 loss not only enhanced the effects of a NF1 deletion by amplifying Ras-driven transcription, but also promoted the gain of H3K27ac following the loss of H3K27me3 (Fig. 2a) [22]. Bromodomain inhibitors, such as JQ1, inhibit the function of enhancers and promoters by competitively interfering with the binding of BRD4 to H3K27ac [53]. Notably, PRC2 loss conferred higher sensitivity to the bromodomain inhibition to both MPNST and JAK2V617F myelofibrosis cells [22, 46]. These studies provide a novel rationale toward a therapeutic strategy for eradicating tumors associated with PRC2 insufficiency.

Mechanism of EZH2 loss-induced epigenetic alterations leading to aberrant transcription of PRC2-target genes. a The loss of EZh2-PRC2 induces an “epigenetic switch” characterized by reduced H3K27me3 followed by enhanced active histone mark of H3K27ac at certain promoter regions, leading to the activation of potential oncogenes that are normally repressed by Ezh2-PRC2. b Ezh1 acts as an oncogene in the absence of Ezh2-PRC2. Ezh1-PRC2 compensates the loss of Ezh2, increasing levels of H3K27me3 at many promoter regions, including developmental regulators, which may contribute to tumorigenesis. c In the absence of Ezh2, a significant portion of PRC2 target genes that lose H3K27me3, including key hematopoietic regulator genes (e.g. Gata2, Gata3, and Nr4a2), acquire DNA hyper-methylation at transcriptional regulatory regions
While Ezh2 loss aberrantly activates expression of certain oncogenes due to reduced H3K27me3 modification in MDS and MPN cells, Ezh1-PRC2 and other epigenetic modifiers have been shown to compensate for Ezh2 loss in silencing Ezh2-target genes [9, 37]. While canonical Ezh2 target genes were significantly de-repressed on deletion of Ezh2 in a mouse MDS model, a substantial portion of these genes became repressed again over time, suggesting a compensatory function for Ezh1 [6]. Given that Ezh1/Ezh2 double knockout mice showed severely compromised function of HSCs, it appears that Ezh1 is essential for the maintenance of Ezh2-deficient HSCs [7, 9]. These findings indicate that EZH1 functions as an oncogene and contributes to the promotion of the diseases with EZH2 dysfunction (Fig. 2b).
Promoter DNA hypermethylation has been shown to silence expression of tumor suppressor genes, consequently promoting tumorigenesis. During the formation of MDS, Ezh2 loss caused reduction of H3K27me3 levels at PRC2 target genes, and led to DNA hypermethylation at promoter regions of a significant portion of PRC2 target genes, such as hematopoietic regulators (e.g. Gata2, Gata3, and Nr4a2) and multiple developmental pathway genes (Fig. 2c) [37, 54]. Decitabine, a hypomethylating agent, attenuated DNA hypermethylation followed by de-repression of target genes expression to some extent, and attenuated the proliferative capacity of MDS cells [54]. These results clearly indicate that alternative epigenetic machineries contribute to the development of MDS in the setting of EZH2 insufficiency.
Early T cell precursor (ETP) ALL has been identified as a new pathologic entity associated with poor outcome in patients with T-ALL. In contrast to cortical T-ALL, which is dominantly driven by NOTCH1-activating mutations, ETP-ALL has been characterized by activating mutations in genes regulating cytokine signaling (e.g. JAK-STAT, RAS) and loss-of-function mutations in PRC2 components, including EZH2, SUZ12, and EED (in total 42.2%) [55]. In mice lacking p16 Ink4a /p19 ARF, Ezh2 loss cooperated with oncogenic N-RAS Q61K to develop ETP-ALL accompanied with enhanced expression of Hoxa9, a potent oncogene of AML, due to reduced H3K27me3 at the promoter region [56]. While these findings are consistent with a tumor-suppressive function of EZH2 in ETP-ALL, it remains unknown whether PRC2 dysfunction drives the formation of ETP-ALL via a mechanism similar to that identified in myeloid malignancies with EZH2 insufficiency.
Collectively, EZH2 loss-of-function mutations compromise regulation of H3K27me3 and induce aberrant gene expression, including both gene activation and silencing, in cooperation with a variety of epigenetic machineries, leading eventually to the development of MDS, MPN, and T-ALL. Pharmacological inhibition of PRC2 function may thus promote tumorigenesis in certain cell contexts, highlighting the need for caution when considering this therapeutic approach.
Conclusion
Since EZH2 dysregulation may be integrated with multiple epigenetic events, depending on the cell context and other coexisting mutations, the role of EZH2 in tumorigenesis is highly diverse among cell types. Its transcriptional impacts are also complicated; its primary effect is repression, but may also function as an activator in complexes with non-PcG proteins or via dynamic genome-wide re-distribution of EZH2 activating mutants. Therefore, it remains important to determine how dysregulated EZH2 promotes tumorigenesis in PRC2-dependent and -independent manners. Pharmacological inhibition of PRC2 oncogenic function in cancers is now being tested extensively in pre-clinical and clinical studies. These studies will provide valuable information on the role of PRC2 in cancer and may lead to the development of better therapeutic approaches specific to individual cancer types. It will also be important to determine the downstream targets of EZH2 that contribute to tumorigenesis and to identify collaborating epigenetic pathways (i.e. epigenetic switch and DNA methylation), as these may provide additional novel therapeutic targets against cancers. Thus, EZH2 has multifaceted functions in cancer, and holds key in tumorigenesis in a wide range of cancers. Precise understanding of the epigenetic alterations caused by EZH2 dysregulation is essential for establishing true epigenetic cancer therapies.
References
Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10(10):697–708.
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–80.
Wang H, Wang L, Erdjument-Bromage H, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431(7010):873–8.
Kondo T, Ito S, Koseki H. Polycomb in transcriptional phase transition of developmental genes. Trends Biochem Sci. 2016;41(1):9–19.
Sashida G, Iwama A. Epigenetic regulation of hematopoiesis. Int J Hematol. 2012;96(4):405–12.
Takamatsu-Ichihara E, Kitabayashi I. The roles of Polycomb group proteins in hematopoietic stem cells and hematological malignancies. Int J Hematol. 2016;103(6):634–42.
Xie H, Xu J, Hsu JH, et al. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014;14(1):68–80.
Hidalgo I, Herrera-Merchan A, Ligos JM, et al. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell. 2012;11(5):649–62.
Mochizuki-Kashio M, Aoyama K, Sashida G, et al. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1-dependent manner. Blood. 2015;126(10):1172–83.
Xu J, Shao Z, Li D, et al. Developmental control of polycomb subunit composition by GATA factors mediates a switch to non-canonical functions. Mol Cell. 2015;57(2):304–16.
Kamminga LM, Bystrykh LV, De Boer A, et al. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006;107(5):2170–9.
Herrera-Merchan A, Arranz L, Ligos JM, et al. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat. Commun. 2012;3:623.
Zhou M, Barrette TR, Kumar-Sinha C, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:388–90.
Bachmann IM, Halvorsen OJ, Collett K, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24(2):268–73.
Bracken AP, Pasini D, Capra M, et al. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22(20):5323–35.
Yu J, Yu J, Mani RS, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17(5):443–54.
Benetatos L, Vartholomatos G, Hatzimichael E. Polycomb group proteins and MYC: the cancer connection. Cell Mol Life Sci. 2014;71(2):257–69.
Sander S, Bullinger L, Klapproth K, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112(10):4202–12.
Lee ST, Li Z, Wu Z, et al. Article context-specific regulation of NF-kB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43(5):798–810.
Xu K, Wu Z, Groner A, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;388:1465–70.
Kim KH, Kim W, Howard TP, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21(12):1491–7.
De Raedt T, Beert E, Pasmant E, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514(7521):247–51.
Wassef M, Rodilla V, Teissandier A, et al. Impaired PRC2 activity promotes transcriptional instability and favors breast tumorigenesis. Genes Dev. 2015;29(24):2547–62.
Kallappagoudar S, Yadav RK, Lowe BR, Partridge JF. Histone H3 mutations-a special role for H3.3 in tumorigenesis? Chromosoma. 2015;124(2):177–89.
Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5.
Yap DB, Chu J, Berg T, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117(8):2451–9.
Sneeringer CJ, Scott MP, Kuntz KW, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci USA. 2010;107(49):20980–5.
McCabe MT, Graves AP, Ganji G, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci USA. 2012;109(8):2989–94.
Su I, Dobenecker M-W, Dickinson E, et al. Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell. 2005;121(3):425–36.
Béguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23(5):677–92.
Souroullas GP, Jeck WR, Parker JS, et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med. 2016;22(6):1–15.
Béguelin W, Teater M, Gearhart MD, et al. EZH2 and BCL6 cooperate to assemble CBX8-BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell. 2016;30(2):197–213.
Young MD, Willson TA, Wakefield MJ, et al. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011;39(17):7415–27.
Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21.
Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–506.
Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30(27):3376–82.
Sashida G, Harada H, Matsui H, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun. 2014;5:4177.
Neff T, Sinha AU, Kluk MJ, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci. 2012;109(13):5028–33.
Tanaka S, Miyagi S, Sashida G, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012;120(5):1107–17.
Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599–612.
Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–6.
Nikoloski G, Langemeijer SMC, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8):665–7.
Kotini AG, Chang C, Boussaad I, et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat Biotechnol. 2015;33(6):646–55.
Guglielmelli P, Biamonte F, Score J, et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood. 2011;118(19):5227–34.
Muto T, Sashida G, Oshima M, et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med. 2013;210(12):2627–39.
Sashida G, Wang C, Tomioka T, et al. The loss of Ezh2 drives the pathogenesis of myelofibrosis and sensitizes tumor-initiating cells to bromodomain inhibition. J Exp Med. 2016;213(8):1459–77.
Shimizu T, Kubovcakova L, Nienhold R, et al. Loss of Ezh2 synergizes with JAK2 -V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J Exp Med. 2016;213(8):1479–96.
Yang Y, Akada H, Nath D, Hutchison RE, Mohi G. Loss of Ezh2 cooperates with Jak2V617F in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood. 2016;127(26):3410–24.
Ntziachristos P, Tsirigos A, Van Vlierberghe P, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012;18(2):298–301.
Simon C, Chagraoui J, Krosl J, et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 2012;26(7):651–6.
Guglielmelli P, Zini R, Bogani C, et al. Molecular profiling of CD34 + cells in idiopathic myelofibrosis identifies a set of disease-associated genes and reveals the clinical significance of Wilms’ tumor gene 1 (WT1). Stem Cells. 2007;25(1):165–73.
Oguro H, Yuan J, Tanaka S, et al. Lethal myelofibrosis induced by Bmi1-deficient hematopoietic cells unveils a tumor suppressor function of the polycomb group genes. J Exp Med. 2012;209(3):445–54.
Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–73.
Hasegawa N, Oshima M, Sashida G, et al. Impact of combinatorial dysfunctions of Tet2 and Ezh2 on the epigenome in the pathogenesis of myelodysplastic syndrome. Leukemia. 2016; in press.
Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–63.
Danis E, Yamauchi T, Echanique K, et al. Ezh2 Controls an Early Hematopoietic Program and Growth and Survival Signaling in Early T Cell Precursor Acute Lymphoblastic Leukemia. Cell Rep. 2016;14(8):1953–65.
Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research (#15H02544, #25130702, #26115002, #23591367, #26461396) and Scientific Research on Innovative Areas “Cell Fate” (#22118004) and “Stem Cell Aging and Disease” (#25115002) from MEXT, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
About this article

Cite this article
Sashida, G., Iwama, A. Multifaceted role of the polycomb-group gene EZH2 in hematological malignancies. Int J Hematol 105, 23–30 (2017). https://doi.org/10.1007/s12185-016-2124-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-2124-x