
Abstract
Cytarabine arabinoside (Ara-C) is the most important agent for treating acute myeloid leukemia (AML). Here, we genotyped 11 single nucleotide polymorphisms (SNPs) of seven Ara-C metabolism-related genes in 39 AML patients who had received high-dose Ara-C as a single-agent treatment. Univariate analysis identified three SNPs that were significantly associated with shorter time-to-relapse (TTR): CTPS rs12144160 GG compared to AA/AG, DCTD rs9990999 AG/GG compared to AA, and SLC29A1 rs693955 CC compared to AA/AC. Multivariate analysis of TTR revealed the SLC29A1 rs693955 CC genotype and first induction failure to be significantly associated with a shorter TTR. The DCTD rs9990999 AG/GG and SLC29A1 rs693955 CC genotypes were also significantly associated with shorter duration of neutropenia. The results of our study suggest that SNP analysis can be an important tool in improving drug responsiveness and enabling a better understanding of this condition and the development of tailor-made treatments for AML patients who benefit from consolidated high-dose Ara-C therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is hematological malignancy characterized by the rapid growth of abnormal white blood cells that accumulate in the bone marrow and interfere with the production of normal blood cells. Treatment strategies for AML that use one of the various cytarabine arabinoside (Ara-C) agents have remained the general choice of clinicians for more than 40 years. Multiple clinical trials have demonstrated complete remission (CR) rates of 50–60 % and overall survival rates of 30–40 % among AML patients receiving such Ara-C-based therapy [1–3]. However, many studies indicate that AML describes a heterogeneous collection of diseases characterized by distinct chromosomal abnormalities and cytogenetic mutations, and as such, the most suitable general treatment for AML is still unclear. Gene variations in leukemic cells significantly associated with prognosis have now been identified, with consequent prognostic improvement [4–7], and we propose that similar improvements in AML treatment could be achieved by better understanding the genetic polymorphisms related to the pharmacokinetics of Ara-C.
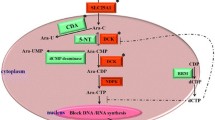
Ara-C is one of the nucleotide-analog therapeutic agents, which are transported into cells by nucleoside transporters including solute carrier family 29 member 1 (SLC29A1) [8]. Intracellular Ara-C is phosphorylated into Ara-C monophosphate (Ara-CMP) by deoxycytidine kinase (DCK) and eventually to Ara-C triphosphate (Ara-CTP), which competes with deoxycytidine triphosphate (dCTP) for incorporation into DNA and subsequent blocking of DNA synthesis causing cell death. In turn, cytidine deaminase (CDA) and deoxycytidylate deaminases (DCTD) catalyze the conversion of Ara-C and Ara-CMP into an inactive form [9], and 5′-nucleotidase cytosolic II (NT5C2) activity opposes that of DCK by dephosphorylating Ara-CMP. In addition, cytidine-5′-triphosphate synthetase (CTPS) and ribonucleotide reductase (RRM1/RRM2) are enzymes that regulate the intracellular CTP/dCTP pools, with exhaustion of the CTP/dCTP pools via facilitated Ara-C phosphorylation causing incorporation of Ara-CTP into DNA by reducing the feedback inhibition of DCK [10] (Fig. 1).

Metabolic pathway of Ara-C. Ara-C is transported into cells by SLC29A1. Intracellular Ara-C is phosphorylated by DCK. Ara-CTP competes with dCTP for incorporation into DNA. CDA catalyzes the conversion of Ara-C into Ara-U, and DCTD catalyzes the conversion of Ara-CMP into Ara-UMP. CTPS catalyzes the conversion of UTP to CTP. Exhaustion of the CTP/dCTP pools due to facilitated Ara-C phosphorylation causes incorporation of Ara-CTP into DNA by reducing the feedback inhibition of DCK. RRM1/RRM2 is an enzyme involved in DNA synthesis. The enzyme regulates intracellular dCTP pools, which in turn, have been implicated in the development of Ara-C resistance
Understanding the pharmacogenetic response to Ara-C could lead to personalized treatment strategies and improved outcomes in AML patients. Indeed, each of the Ara-C metabolism-associated genes exhibits a significant degree of genetic variation, particularly via single nucleotide polymorphisms (SNPs), and several studies of individual SNPs in Ara-C metabolic genes have reported that genetic background plays an important role in the clinical outcomes of AML patients receiving Ara-C-based therapy [11–13]. In one such study, Gamazon et al. [11] conducted a meta-analysis of genome-wide association studies involving 523 lymphoblastoid cell lines from individuals of European, African, Asian, and African American ancestry, and identified 18 of 33 SNPs associated with either cytarabine 50 % inhibitory concentration in leukemia cells or clinical response parameters among patients randomized to receive low-dose or high-dose Ara-C plus daunorubicin and etoposide. In addition, Kim et al. [13] reported that the SLC29A1 rs3734703 AA/AC genotype in combination with TYMS rs2612100 AA genotype was significantly associated with shorter relapse-free survival in Korean AML patients received an induction regimen of Ara-C and idarubicin followed by sequential consolidation therapy with Ara-C and anthracyclines or hematopoietic stem cell transplantation (HSCT). However, in these reports, the patient background included anthracycline agents or HSCT and thus might not accurately reflect the influence of genetic polymorphism on Ara-C metabolism.
Analyzing the combined effects of SNPs may provide evidence of drug response. Accordingly, we hypothesized that sensitivity to Ara-C could be influenced by SNP located in Ara-C metabolic genes and thus focused on high-dose Ara-C as single-agent therapy. In this study, we simply examined the association between SNPs in such genes and the clinical outcome of AML patients receiving high-dose Ara-C without HSCT.
Materials and methods
Study patients
We selected de novo AML patients who received high-dose Ara-C as consolidation therapy, and whose bone marrow or peripheral blood samples were stored in our laboratory. We excluded one patient diagnosed as M3 subtype and one patient who received HSCT in the first CR. All subjects enrolled in this study provided informed consent for genetic analysis. This study was approved by the Institutional Review Board of Tokai University Hospital.
SNP selection
Seven Ara-C metabolic genes, CDA, CTPS, DCK, DCTD, NT5C2, RRM1 and SLC29A1, were evaluated. SNPs were selected based on SNP frequency data from the International Hap-Map project (http://hapmap.ncbi.nlm.nih.gov/) and The National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/). SNP frequency data were identified according to the HapMap Genome Browser release #27 (Phase 1, 2 and 3—merged genotypes and frequencies) of Japanese in Tokyo, Japan (JPT) database to estimate linkage disequilibrium (LD) blocks by Haploview software (version 4.2) [14]. The criteria for SNP selection were thus as follows: (1) minor allele frequency (MAF) >0.25 in JPT; (2) one or two SNPs were selected from each evaluated gene; (3) SNP reported by previous study [15], or located with each different LD blocks of the highest MAF. Allele frequency data of rs9937 was lacking in the JPT, so we instead search SNP frequency data of the CHB + JPT data in NCBI. Finally, eleven SNPs (CDA; rs10916827, rs477155, CTPS; rs4132440, rs12144160, DCK; rs12648166 DCTD; rs9542, rs9990999, NT5C2; rs3736922, RRM1; rs9937, SLC29A1; rs693955, 9394992) in each of the gene locus were selected.
SNP genotyping
Genomic DNA was isolated from bone marrow or peripheral blood individually using a QIAamp mini DNA kit (Qiagen, Valencia, CA, USA). SNP genotyping was performed using the TaqMan platform in 96-well plates and read with the Sequence Detection Software on a 7500 Real-Time PCR System according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA, USA). Primers and probes were supplied by Applied Biosystems.
Definition of clinical response and hematological toxicity
All clinical information about the patients was obtained from our institution database.
Overall survival (OS) was measured from the date of diagnosis to the date of death from any cause. Time to relapse (TTR) was measured from the date of diagnosis to the date of relapse. Relapse was defined as the presence of more than 5 % blast cells in the bone marrow or reappearance of blast cells in the peripheral blood. Hematological toxicity was measured by duration of neutropenia and thrombocytopenia during each high-dose Ara-C course. Neutropenia was defined as an absolute neutrophil count less than 500/μl. Thrombocytopenia was defined as an absolute platelet count less than 50,000/μl.
Statistical analysis
For SNP analysis, we tested two genetic models: the dominant model (major allele homozygous + heterozygous vs. minor allele homozygous) and the recessive model (major allele homozygous vs. heterozygous + minor allele homozygous). For univariate analysis, OS probabilities were estimated by the Kaplan–Meier method, and differences in the distributions between the dominant and recessive model of each SNP were evaluated using the log-rank test. TTR was estimated by the cumulative incidence method, and Gray’s test was used to compare distribution differences between the genotypes [16]. Death without experiencing a relapse was considered a competing event with experiencing first relapse.
For multivariate analysis, the Fine–Gray regression model [5] was used for the sub-distribution hazard of a competing risk to analyze the effect of baseline risk factors on the cumulative incidence function of relapse. Factors associated with a two-side P value of less than 0.05 in the univariate SNP analysis and known prognostic risk factors for the Japanese population [17], such as age >50 years, performance status >2, myeloperoxidase (MPO) positivity of blasts >50 %, and first induction failure, were included in the multivariate analysis. We used a stepwise regression based on P values and retained only the statistically significant variables in the final model. This analysis did not include the factor of the DCTD rs9990999 AA genotype, because no patients with DCTD rs9990999 AA genotype experienced relapse. Thus, the DCTD rs9990999 AA genotype could not be treated as a single multivariate factor for the time-to-event analysis. For hematological toxicity analysis, comparisons between the dominant and recessive models of each SNP were performed using Student’s t test.
All statistical analyses were performed with EZR (Saitama Medical Center, Jichi Medical University), which is a graphical user interface for R (The R Foundation for Statistical Computing, version 2.13.2) [18]. More precisely, it is a modified version of R commander (version 1.6–3) that includes statistical functions frequently used in biostatistics. For all analyses, P values were two-tailed, and a P value of less than 0.05 was considered significant.
Results
Patient characteristics and treatments
Thirty-nine AML patients were eligible for this study. Table 1 summarizes the patients’ characteristics. The median age of patients was 54.0 years (range 23.0–71.0 years) and the male/female proportion was 26/13. The most frequent French–American–British (FAB) subtype was M2 (56.4 %) followed by M1 (20.5 %). M0, M6, and M7 subtypes were not represented in this population. In total, 14 patients had a good cytogenetic risk based on karyotype; 12 patients had t(8; 21) (q22;q22) and 2 patients had inv(16) (p13q22). Another 4 patients had a poor cytogenetic risk; 2 patients had del(7) and 2 patient had a complex karyotype. A total of 14 patients (35.9 %) were of normal karyotype, and 7 patients had an unspecified karyotype. Three of the 19 patients in whom the FLT3 internal tandem duplication (ITD) mutation information was identified had a FLT3/ITD mutation; however, the FLT3/ITD mutation was not examined for the other 20 patients and data on other molecular abnormalities were not available. Median white blood cell (WBC) count at diagnosis was 20,800/µl (range 900–47,4800/µl). Eight patients had received their first induction regimen consisting of Ara-C with daunorubicin, and another 31 patients received an induction regimen consisting of Ara-C with idarubicin. Thirty-three patients (84.6 %) achieved CR after the first induction regimen, and six patients (15.4 %) needed two or more induction regimens to achieve CR. Once patients achieved complete remission, the patients received consolidation therapy consisting of high-dose Ara-C. Seventeen patients received three courses of Ara-C dose of 2.0 g/m2 for 5 days. Two patients received two courses of Ara-C dose of 2.0 g/m2 for 5 days and one patient was died in the second course of consolidation therapy due to severe infection. Three patients died in the first course. Other patients received various doses and durations of Ara-C treatment according to the physician’s clinical decision, as follows: one patient received three courses of Ara-C dose of 3.0 g/m2 for 5 days; one patient received three courses of Ara-C dose of 3.0 g/m2 for 4 days; two patients received four courses of Ara-C dose of 3.0 g/m2 for 3 days; one patient received two courses and two patients received three of Ara-C dose of 2.0 g/m2 for 3 days; and, one patient received two courses and four patients received three of Ara-C dose of 1.0 g/m2 for 5 days. Nineteen patients (48.7 %) relapsed during the follow-up period and the median TTR was 306 days (range 152–1271 days). The median follow-up period overall survival was 833 days (range 55–3931 days).
Results of SNP genotyping
All SNP genotyping was successful among the AML patients, as summarized in Table 2.
The SNP genotypes could be divided into three groups, and comparison between groups only was analyzed statistically, due to insufficient minor allele frequency.
SNP effect on treatment outcomes
The effects of the two SNP genetic models on OS and TTR are summarized in Table 3. In the univariate analysis, no SNP was a significant prognostic factor for OS. However, three SNPs, individually, had associations with TTR (Fig. 2). The CTPS rs12144160 GG genotype was significantly associated with shorter TTR compared to the AA/AG genotype (2-year relapse rate 0.694 [95 % CI 0.258–0.907] vs. 0.363 [95 %CI 0.177–0.552], P = 0.0209). The DCTD rs9990999 AG/GG genotype was significantly associated with shorter TTR compared to the AA genotype (2-year relapse rate 0.529 [95 % CI 0.331–0.692] vs. NA [NA–NA], P = 0.0255). The SLC29A1 rs693955 CC genotype was significantly associated with shorter TTR compared to the AA/AC genotype (2-year relapse rate 0.683 [95 % CI 0.416–0.848] vs. 0.131 [0.019-0.353], P = 0.00261). There were 14 patients with CBF leukemia and 21 patients with intermediate risk group. The SLC29A1 rs693955 CC genotype with intermediate risk (n = 14) was significantly associated with shorter TTR compared to the AA/AC genotype (n = 7) (2-year relapse rate 0.701 [95 % CI 0.295–0.902] vs. 0.214 [0.002–0.689], P = 0.0498). There was no statistical significance of the comparison for other SNPs and TTR regarding cytogenetic risk groups.

Significant effect of SNP on time to relapse. Time to relapse was estimated by the cumulative incidence method, and Gray’s test was used to compare differences between genotypes with respect to the SNP distributions. Death without experiencing a relapse was considered a competing event with experiencing first relapse. a SNP effect of CTPS rs12144160. b SNP effect of DCTD rs9990999. c SNP effect of SLC29A1 rs693955
Multivariate analysis of TTR revealed that the SLC29A1 rs693955 CC genotype (HR 7.659 [95 % CI 1.98–29.63], P = 0.0096) and first induction failure (HR 3.613 [95 % CI 1.37–9.55]) were significantly associated with shorter TTR (Table 4).
SNP effect on hematological toxicity
The total number of high-dose Ara-C was 109, and febrile neutropenia (FN) observed in 83 cases including 4 mortalities due to exacerbation of infections. The dead cases were excluded from the duration analysis because they did not recover from the neutropenia or the thrombocytopenia. The duration of hematological toxicity analysis was therefore analyzed for 105 treatment courses. The mean durations of neutropenia and thrombocytopenia were 14.1 days (95 % confidential interval [CI]; 12.8–15.4 days) and 14.3 days (95 %CI; 12.6–16.0 days), respectively. Administration of granulocyte colony-stimulating factor (GCSF) was noted in 55/109 courses. Table 5 summarized the statistical analysis of SNPs compared to duration of neutropenia or thrombocytopenia. Figure 3 shows the three candidate SNPs and the association with duration of neutropenia. The DCTD rs9990999 AG/GG genotype was significantly associated with shorter duration of neutropenia compared to the AA genotype (13.2 ± 5.77 vs. 19.5 ± 9.11 days, P = 0.000497). The SLC29A1 rs693955 CC genotype was significantly associated with shorter duration of neutropenia compared to the AA/AC genotype (13.1 ± 5.69 vs. 15.8 ± 7.87 days, P = 0.0386) and also thrombocytopenia (12.7 ± 7.89 vs. 16.9 ± 9.15 days, P = 0.0116). The frequency of GCSF administration was 1 in 16 of the DCTD rs9990999 AA genotypes and 54 in 93 of AG/GG genotypes, and was similar between the SLC29A1 AA/AC genotype (20/42) and CC genotype (32/67). The SLC29A1 AA/AC genotype required longer duration of GCSF administration than CC genotype (16.1 ± 5.54 vs. 13.1 ± 4.58 days, P = 0.0377). Concerning neutropenia without GCSF, the DCTD rs9990999 AG/GG genotype (n = 39) was significantly associated with shorter duration of neutropenia compared to the AA genotype (n = 15) (15.1 ± 5.44 vs. 19.5 ± 9.11 days, P = 0.0314), and the SLC29A1 rs693955 CC genotype (n = 32) was significantly associated with shorter duration of neutropenia compared to the AA/AC genotype (n = 22) (14.8 ± 5.33 vs. 18.5 ± 5.27 days, P = 0.0468). The frequency of FN was 12 of 16 DCTD rs9990999 AA genotypes, 71 of 93 AG/GG genotypes, 30 of 42 SLC29A1 AA/AC genotypes, and 53 of 67 CC genotypes. In addition, the four cases that died during high dose Ara-C treatment all had the same SLC29A1 rs693955 AA/AC genotype. However, there was no severe acute neurotoxicity caused by high-dose Ara-C therapy observed in the patients, and other acute, severe non-hematologic side effects were not documented in the available clinical information.

Duration of neutropenia. Three SNPs associated with TTR are shown in this figure. a SNP effect of CTPS rs12144160. b SNP effect of DCTD rs9990999. c SNP effect of SLC29A1 rs693955. CTPS rs12144160 was not associated with hematological toxicity. The DCTD rs9990999 AA genotype was associated with a longer duration of neutropenia than the AA or CC genotype. The SLC29A1 rs693955 AA genotype was associated with a longer duration of both neutropenia and thrombocytopenia than the AA or CC genotype
Discussion
The present study suggested that the three SNPs, rs12144160 in the CTPS gene, rs9990999 in the DCTD gene, and rs693955 in the SCL29A1 gene could influence outcomes in AML patients receiving high-dose Ara-C treatment.
The catalytic conversion of UTP to CTP is accomplished by the CTPS enzyme encoded by CTPS, and increased Ara-C sensitivity results from decreased CTP/dCTP pools caused by inhibition of CTPS with cyclopentenyl cytosine in myeloid leukemia and T-lymphoblastic leukemia cell lines [19, 20]. Although Ara-C resistance caused by clustered mutations within the coding region of CTPS have been identified in Chinese hamster ovary cells, no mutations were identified in the regions indicated from recurrent or resistant acute leukemia in 36 patients [10]. In our study, the CTPS rs12144160 GG genotype was significantly associated with shorter TTR compared to the AA/AG genotype, suggesting that the GG genotype patients had higher levels of CTPS expression or activity than those with the AA/AG genotype. However, rs12144160 is located in an intronic region and therefore might affect RNA expression rather than enzyme activity.
SNP rs9990999 in the DCTD gene was a significant prognostic factor for TTR in this study, and the AG/GG genotype was significantly associated with shorter TTR compared to the AA genotypes. Interestingly, the patient with an AA genotype showed a long duration of neutropenia and did not experience relapse in our study. The protein encoded by the DCTD gene catalyzes the deamination of Ara-CMP to Ara-UMP and is allosterically activated by dCTP and inhibited by dTTP. Schröder et al. [9] reported that the expression level of DCTD was not associated with Ara-C sensitivity, while a non-synonymous SNP, the A172G mutation causing Asn58Asp, on the coding regions and causing loss of activity for gemcitabine monophosphate was identified in Caucasian and African ethnic groups in vitro assays [21], although the minor allele frequency was too low for meaningful association analysis with clinical response to Ara-C in this previous study [12]. However, nonsynonymous SNPs, including A172G, have not been observed in the DCTD gene in a Japanese population. There is also no evidence of functional SNPs in the DCTD gene, although our findings showed some kind of gene function for rs9990999 in TTR and neutrophil toxicity.
Our univariate and multivariate analysis found that the SLC29A1 rs693955 CC genotype was significantly associated with shorter TTR and shorter duration of hematological toxicity. The SLC29A1 gene encodes the human equivalent of nucleoside transporter 1 (hENT1), a protein that resides in the plasma membrane to mediate the cellular uptake of cytotoxic nucleosides as Ara-C from the surrounding medium. Although multiple alternatively spliced variants have been found for the SLC29A1 gene, they all encode the same protein, and thus a deficiency in hENT1 expression might be the basis of cellular resistance to Ara-C [22]. SNPs have been previously detected in the SLC29A1 gene from Japanese populations and some SNPs have been implicated in the efficacy of Ara-C [23] and the mRNA expression [24, 25]. However, hENT1 is a limiting determinant of Ara-C efficacy, and the simple diffusion rate of Ara-C exceeds its pump-mediated transport in high plasma concentrations of Ara-C [26]. Although it is less likely that Ara-C is taken up into the cell by SLC29A1 at the 50-µM plasma concentration reached by high-dose Ara-C, rs693955 located in the SLC29A1 gene might still influence clinical outcomes, based on our multivariate analysis identifying first induction failure and rs693955 CC genotype as independent prognostic factors. Suzuki et al. [25] reported that mRNA levels in the rs6932345 wild-type (A>C) and rs747199 wild-type (G>C) were higher than in the mutation carriers, and LD block analysis from the HapMap database linked the rs693955 C allele with the rs747199 C allele at a frequency of 14.8 %. Conversely, the rs693955 A allele was not linked with the rs747199 G allele. Thus, we proposed that patients with the rs693955 CC genotype have lower expression levels of hENT1 and consequently, shorter TTR and lower hematological toxicity. In addition, Pérez-Torras et al. [27] reported that overexpression of hENT1 in a relatively low transporter activity background increased the uridine uptake, resulting in a decreased amount of mRNA encoding key nucleotide metabolism enzymes, such as DCK and RR, and reduced cell cycle progression in the cell lines derived from human pancreatic adenocarcinomas. Nucleotide metabolism with the rs693955 CC genotype might be easy to change by the similar action of hENT1 overexpression. High intracellular concentrations of Ara-C or the product of Ara-C metabolism due to high-dose Ara-C therapy may have caused the decrease in nucleotide metabolism enzymes and cell cycle progression. The rs693955 CC genotype patient in this study might therefore have a phenotypic resistance to Ara-C, leading to the rapid recovery from cytopenia and the early relapse.
DCK is required for the first phosphorylation of deoxyribonucleosides and Ara-C is the most important enzyme in the activation pathway of Ara-C. However, in our study, the DCK rs12648166 was not associated with therapeutic outcomes. Our study does not include all SNPs on the DCK locus, but only one SNP was available based on our SNP selection criteria, and it is possible other SNPs on the DCK locus with a lower MAF might be functional polymorphisms.
Interestingly, the DCTD rs9990999 AG/GG and SLC29A1 rs693955 CC were associated with shorter time to relapse and shorter duration of neutropenia. This finding suggested that the sensitivity to Ara-C of de novo leukemic cells is not very different from the sensitivity to Ara-C of normal hematopoietic stem cells. Braess et al. [28] reported CDA activities and Ara-C deamination in a variety of the most commonly used leukemic cell lines, fresh blasts, and normal bone marrow cells. However, the cell lines herein had different CDA activity profiles and degrade between 18.5 and 96.5 % of Ara-C to Ara-U. Formation of Ara-CTP is therefore significantly influenced by the differences in cell type-dependent cytidine deaminase activity. In contrast to the cell lines, fresh leukemic blasts and normal bone marrow mononuclear cells show low Ara-C degradation, and cultured cell lines are exposed to unknown selective pressure during a year or even several decades. Therefore, the biological reactions of cultured cells to Ara-C exposure seem to be relatively changed from the primary source leukemia cells. Conversely, the de novo leukemic cell without exposure to Ara-C might maintain a similar Ara-C metabolism to normal cells.
The results of our study suggested that SNP analysis could lead to better drug responsiveness and improved treatments for AML patients who benefit by receiving consolidation therapy with high-dose Ara-C. To our knowledge, this is the first report showing the relationship between SNPs and clinical outcomes in AML patient receiving high-dose Ara-C as single-agent therapy. Previous studies investigated the relationships between SNPs located on Ara-C metabolic genes and clinical response or toxicity with various doses of Ara-C based therapy including hematopoietic stem cell transplantation. RRM1 rs1042919 and rs1561876 were related to intracellular Ara-CTP concentration, CR rate, and OS [29]. CDA rs2072671 and rs532545 were related to OS in a FLT3-ITD mutation-positive normal karyotype AML patient, and NT5C2 rs10883841 was related to OS in a FLT3-ITD mutation-negative case [30]. In addition, the SLC29A1 rs3734703 AA/AC genotype in combination with TYMS rs2612100 was significantly associated with relapse-free survival and DCK rs469436 was associated with OS in AML patients [13]. Nevertheless, few studies have determined SNP functions, and it is possible that CDA rs2072671 caused CDA protein variants (p.Lys27Gln) that may be related to the loss of CDA activity [31]. Our study did not include such SNPs because their MAF was lower than 0.25. For particular SNPs to be extracted as therapeutic surrogate markers, the functional meaning of these candidate SNPs must be determined and validated in future cohort studies. It might also be applicable to improve other nucleotide analog treatments which have similar pharmacokinetics system of Ara-C, such as gemcitabine, fluorouracil, and azacitidine by conducting SNP analysis associated with the Ara-C pharmacokinetics. However, our study was limited due to the small sample size and the inherent selection bias, since the patients examined here were already in the first remission state and were receiving high-dose Ara-C as a consolidation therapy. All the meaningful SNPs in our study were in introns. We could therefore not explore the SNP functions based on RNA expression, elongation, or splicing variants of the gene. It is therefore necessary to identify functional SNPs that could be related to LD with the SNPs in our study by genome sequencing and thereby confirm the differences by RNA expression level or enzyme activity. Further studies are therefore needed to reveal the SNP functions and validation cohort studies are warranted.
References
Kumar CC. Genetic abnormalities and challenges in the treatment of acute myeloid leukemia. Genes Cancer. 2011;2:95–107.
Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, European LeukemiaNet, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–74.
Miyawaki S, Ohtake S, Fujisawa S, Kiyoi H, Shinagawa K, Usui N, et al. A randomized comparison of 4 courses of standard-dose multiagent chemotherapy versus 3 courses of high-dose cytarabine alone in postremission therapy for acute myeloid leukemia in adults: the JALSG AML201 Study. Blood. 2011;117:2366–72.
Bullinger L, Döhner K, Bair E, Fröhling S, Schlenk RF, Tibshirani R, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–16.
Sakamaki H, Miyawaki S, Ohtake S, Emi N, Yagasaki F, Mitani K, et al. Allogeneic stem cell transplantation versus chemotherapy as post-remission therapy for intermediate or poor risk adult acute myeloid leukemia: results of the JALSG AML97 study. Int J Hematol. 2010;91:284–92.
Naoe T, Kiyoi H. Gene mutations of acute myeloid leukemia in the genome era. Int J Hematol. 2013;97:165–74.
Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–60.
Wiley JS, Jones SP, Sawyer WH, Paterson AR. Cytosine arabinoside influx and nucleoside transport sites in acute leukemia. J Clin Investig. 1982;69:479–89.
Schröder JK, Seidelmann M, Kirch HC, Seeber S, Schütte J. Assessment of resistance induction to cytosine arabinoside following transfer and overexpression of the deoxycytidylate deaminase gene in vitro. Leuk Res. 1998;22:619–24.
Whelan J, Smith T, Phear G, Rohatiner A, Lister A, Meuth M. Resistance to cytosine arabinoside in acute leukemia: the significance of mutations in CTP synthetase. Leukemia. 1994;8:264–5.
Gamazon ER, Lamba JK, Pounds S, Stark AL, Wheeler HE, Cao X, et al. Comprehensive genetic analysis of cytarabine sensitivity in a cell-based model identifies polymorphisms associated with outcome in AML patients. Blood. 2013;121:4366–76.
Mahlknecht U, Dransfeld CL, Bulut N, Kramer M, Thiede C, Ehninger G, et al. SNP analyses in cytarabine metabolizing enzymes in AML patients and their impact on treatment response and patient survival: identification of CDA SNP C-451T as an independent prognostic parameter for survival. Leukemia. 2009;23:1929–32.
Kim KI, Huh IS, Kim IW, Park T, Ahn KS, Yoon SS, et al. Combined interaction of multi-locus genetic polymorphisms in cytarabine arabinoside metabolic pathway on clinical outcomes in adult acute myeloid leukaemia (AML) patients. Eur J Cancer. 2013;49:403–10.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5.
Rha SY, Jeung HC, Choi YH, Yang WI, Yoo JH, Kim BS, et al. An association between RRM1 haplotype and gemcitabine-induced neutropenia in breast cancer patients. Oncologist. 2007;12:622–30.
Gray RJ. A class of k-sample tests for comparing the cumulative incidence of a competing risk. Annal Statist. 1988;16:1141–54.
Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. Am Statist Assoc. 1999;94:496–509.
Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48:452–8.
Verschuur AC, van Gennip AH, Leen R, Voûte PA, van Kuilenburg AB. Cyclopentenyl cytosine increases the phosphorylation and incorporation into dna of arabinofu-ranosyl cytosine in a myeloid leukemic cell-line. Adv Exp Med Biol. 2000;486:311–7.
Verschuur AC, Brinkman J, Van Gennip AH, Leen R, Vet RJ, Evers LM, et al. Cyclopentenyl cytosine induces apoptosis and increases cytarabine-induced apoptosis in a T-lymphoblastic leukemic cell-line. Leuk Res. 2001;25:891–900.
Gilbert JA, Salavaggione OE, Ji Y, Pelleymounter LL, Eckloff BW, Wieben ED, et al. Gemcitabine pharmacogenomics: cytidine deaminase and deoxycytidylate deaminase gene resequencing and functional genomics. Clin Cancer Res. 2006;12:1794–803.
Gati WP, Paterson AR, Larratt LM, Turner AR, Belch AR. Sensitivity of acute leukemia cells to cytarabine is a correlate of cellular es nucleoside transporter site content measured by flow cytometry with SAENTA-fluorescein. Blood. 1997;90:346–53.
Kim SR, Saito Y, Maekawa K, Sugiyama E, Kaniwa N, Ueno H, et al. Thirty novel genetic variations in the SLC29A1 gene encoding human equilibrative nucleoside transporter 1 (hENT1). Drug Metab Pharmacokinet. 2006;21:248–56.
Suzuki Y, Homma M, Abei M, Hyodo I, Kohda Y. Impact of solute carrier family 29 member 1 (SLC29A1) single nucleotide polymorphisms on mRNA expression in peripheral blood mononuclear cells. Biol Pharm Bull. 2013;36:144–6.
Myers SN, Goyal RK, Roy JD, Fairfull LD, Wilson JW, Ferrell RE. Functional single nucleotide polymorphism haplotypes in the human equilibrative nucleoside transporter 1. Pharmacogenet Genom. 2006;16:315–20.
Capizzi RL, Yang JL, Cheng E, Bjornsson T, Sahasrabudhe D, Tan RS, et al. Alteration of the pharmacokinetics of high-dose ara-C by its metabolite, high ara-U in patients with acute leukemia. J Clin Oncol. 1983;1:763–71.
Pérez-Torras S, García-Manteiga J, Mercadé E, Casado FJ, Carbó N, Pastor-Anglada M, et al. Adenoviral-mediated overexpression of human equilibrative nucleoside transporter 1 (hENT1) enhances gemcitabine response in human pancreatic cancer. Biochem Pharmacol. 2008;76:322–9.
Braess J, Pförtner J, Kern W, Hiddemann W, Schleyer E. Cytidine deaminase—the methodological relevance of AraC deamination for ex vivo experiments using cultured cell lines, fresh leukemic blasts, and normal bone marrow cells. Ann Hematol. 1999;78:514–20.
Cao X, Mitra AK, Pounds S, Crews KR, Gandhi V, Plunkett W, et al. RRM1 and RRM2 pharmacogenetics: association with phenotypes in HapMap cell lines and acute myeloid leukemia patients. Pharmacogenomics. 2013;14:1449–66.
Falk IJ, Fyrberg A, Paul E, Nahi H, Hermanson M, Rosenquist R, et al. Decreased survival in normal karyotype AML with single-nucleotide polymorphisms in genes encoding the AraC metabolizing enzymes cytidine deaminase and 5′-nucleotidase. Am J Hematol. 2013;88:1001–6.
Carpi FM, Vincenzetti S, Ubaldi J, Pucciarelli S, Polzonetti V, et al. CDA gene polymorphisms and enzyme activity: genotype–phenotype relationship in an Italian-Caucasian population. Pharmacogenomics. 2013;14:769–81.
Acknowledgments
We thank the all physicians and nurses at the Hematology and Oncology Department of Tokai University Hospital and laboratory staff in the Department of Hematology and Oncology at Tokai University for their kind support; particularly T. I., E. M., and N. S. for assitance with the genotyping. We also appreciate valuable advice regarding this study from the Morishima research group on Allergic Disease and Immunology from the Ministry of Health, Labor, and Welfare of Japan.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Amaki, J., Onizuka, M., Ohmachi, K. et al. Single nucleotide polymorphisms of cytarabine metabolic genes influence clinical outcome in acute myeloid leukemia patients receiving high-dose cytarabine therapy. Int J Hematol 101, 543–553 (2015). https://doi.org/10.1007/s12185-015-1766-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-015-1766-4