
Abstract
Multidrug resistance (MDR) to chemotherapeutic agents is a major obstacle to curative treatment of cancer. In various types of cancers, overexpression of glucosylceramide synthase (GCS) has been observed to be associated with MDR, thus making GCS a target for reversal of resistance. Our previous work demonstrated that GCS and Bcl-2 are co-overexpressed in the K562/A02 leukemia multidrug-resistant cell line compared with its sensitive counterpart, K562. In the present study, we investigated the effects of GCS on apoptosis in K562/A02 and the associated molecular mechanisms. Our results indicate that the inhibition of GCS caused downregulation of Bcl-2 as well as apoptosis enhancement in response to ADM via the ERK pathway, while JNK or p38 MAPK signaling appeared to play less significant roles in the regulation of apoptosis and MDR in K562/A02 cells. Targeting GCS by siRNA also enhanced ceramide accumulation, which is involved in GCS knockdown-induced inhibition of ERK activation and Bcl-2 expression levels.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multidrug resistance (MDR) to chemotherapeutic agents is a principal problem in the treatment of cancer. It not only limits the effectiveness of chemotherapy but also has side effects on normal tissues. It has been demonstrated that abnormal glucosylceramide synthase (GCS) metabolism is highly associated with MDR. Several reports have shown that GCS upregulates MDR1 expression thereby confers cell resistance by increasing MDR1-mediated drug efflux [1, 2]. However, even if anti-cancer drugs reach their sites of action by passing drug efflux systems, some cells still exhibit resistance via inhibition of pro-apoptotic signaling [3], suggesting that dysregulation of apoptotic signaling plays a very important role in MDR. Previous studies done in our lab demonstrated that both GCS and Bcl-2 gene were overexpressed in K562/A02 drug-resistant cells [4, 5], as well as the clinic chemoresistant cases with leukemia [4]. The Bcl-2 expression was significantly downregulated after GCS knockdown. However, mechanisms of GCS-induced Bcl-2 alteration in drug resistance are poorly understood.
It is known that GCS is a glycosyltransferase in sphingolipid metabolism. It transfers a glucose residue from UDP-glucose to ceramide for glucosylceramide (GlcCer), thus eliminating ceramide and protecting cancers from chemotherapeutics. Since ceramide generated in response to diverse stress factors is implicated as an inducer of cellular growth inhibition and apoptosis, we hypothesize that the alteration of ceramide accumulation might be one of the mechanisms of GCS-induced Bcl-2 overexpression.
In addition, earlier work from our group revealed that the inhibitory action of GCS knockdown in cell resistance to drugs involved inhibition of ERK cascade [6]. Herein, we showed that knockdown of GCS-induced Bcl-2 downregulation through inhibition of ERK signaling. Endogenous formation of ceramide by targeting GCS using siRNA was the mechanism contributing to ERK cascade and Bcl-2 inhibition.
Materials and methods
Materials
Chemicals and reagents used in this study included: RPMI1640 growth medium and penicillin–streptomycin (Hyclone, Logan, UT, USA); fetal bovine serum (FBS) (Sigma–Aldrich, St. Louis, MO, USA); Cell Counting Kit-8 (Dojindo, Kumamoto, Japan); adriamycin (ADM) (Shanghai Hua Lian Co., Ltd., Shanghai, China); U0126 (MEK-ERK pathway inhibitor) (Cell Signaling Technology, Beverly, MA, USA); C6-Ceramide (N-hexanoyl-d-erythro-sphingosine) (Matreya Inc., Pleasant Gap, PA, USA); PVDF membrane and Chemiluminescent HRP Substrate (Millipore, Bedford, MA, USA).
Cell culture
The drug-resistant K562/A02 human chronic leukemia cell line was purchased from Hematology Institute of Chinese Academy of Medical Sciences in Tianjin. Cells were grown and maintained in RPMI1640 growth medium supplemented with 10 % (v/v) heat-inactivated FBS, 100 units/mL penicillin and 100 μg/mL streptomycin at 37 °C in a 5 % CO2 humidified atmosphere. K562/A02 cells were routinely maintained in medium containing 5 μg/mL ADM to ensure the drug-resistant phenotype and incubated in drug-free medium for 2 weeks prior to experiments. Cells were fed with fresh medium every 3 days and used for experiments in log phase.
Knockdown of gene expression using siRNA
The siRNA oligonucleotides against GCS were selected according to previous studies [2, 5] in our laboratory and synthesized by Shanghai Gene Pharma Co., Ltd. (Shanghai, China). The siRNA sequences used were as follows: GCS siRNA: 5′-GGAUUAUCCCAAAUAUGAATT-3′ and the scrambled siRNA used as a negative control: 5′-UUCUCCGAACGUGUCACGUTT-3′. Before transfection, K562/A02 cells were seeded in 24-well plates and maintained in medium until cell confluence reached nearly 60 %. A total of 100 nM siRNA were transfected into K562/A02 cells as previously described [7]. After a 48-h incubation period, the inference efficiency was analyzed by real-time PCR.
Annexin-V-FITC/PI detection of apoptotic cells
Apoptotic cells were detected using Annexin-V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit (eBioscience, San Diego, CA, USA). Cells were harvested and washed with PBS, and then stained with Annexin-V/PI according to the manufacturer’s recommendation. Stained samples were analyzed by flow cytometry (FCM) (FACSCanto II, BD Biosciences, San Diego, CA, USA) with excitation and emission setting of 488 and 530 nm, respectively. At least 2 × 104 cells were counted in each sample.
Cell proliferation assay
Cell proliferation was examined by CCK-8 kit. Briefly, 5 × 103 cells/well in 100 μL medium were seeded in 96-well culture plates and exposed to indicated concentrations of ADM for 72 h. 10 μL of Cell Counting Kit-8 reagent was added to each well and incubated for another 3 h at 37 °C. The absorbance of wells was then read at 450 nm using a spectrophotometer. The IC50 value (defined as the concentration of drug that inhibited cell growth by 50 %) for K562/A02 cell line to chemotherapeutic agent was determined.
Ceramide measurement
Endogenous ceramides were measured through normal phase high-performance liquid chromatography coupled atmospheric pressure chemical ionization–mass spectrometry (LC/MS) as described previously [8]. The ceramide levels were normalized against inorganic phosphorus (Pi) in cells.
Real-time PCR
Total RNAs were extracted by Trizol reagent (Invitrogen, Carlsbad, CA, USA) and reversely transcribed with Reverse Transcription System (Promega, Madison, WI, USA). cDNA was amplified using Premix Ex TaqTM kit (Takara, Dalian, China) and analyzed by Light Cycler PCR instrument (Roche Co.). GAPDH gene was performed as an endogenous control to normalize the mRNA levels of each sample. The relative value of each cDNA was analyzed by △△Ct relative quantitative analysis method. Oligonucleotides sequences of upstream and downstream primers were as follows: GCS F: 5′-TGCTCAGTACATTGCCGAAGA-3′, GCS R: 5′-TGGACATTGCAAACCTCCAA-3′ and GCS probe: 5′-TTATGGCCAAAGCGATAGCTGACCGAG-3′; Bcl-2 F: 5′-GGATCATGCTGTACTTAA-3′, Bcl-2 R: 5′-TGAGGCACGTTATTATTAG-3′ and Bcl-2 probe: 5′-CAGTCTACTTCCTCTGTGATGTTGT-3′; GAPDH F: 5′-CCCATGTTCGTCATGGGTGT-3′, GAPDH R: 5′-TGGTCAGAGTCCTTCCACGATA-3′ and GAPDH probe: 5′-CTGCACCACCAACTCTTAGCACCC-3′. Individual test was carried out in triplicate.
Western blotting analysis
Cells were harvested by centrifugation, washed with PBS and lysed in RIPA buffer (Beyotime, Beijing, China). Total protein concentration was measured using a BCA protein determination kit (Beyotime, Beijing, China). Equal aliquots of protein were separated by 10 % sodium dodecylsulfate-polyacrylamide gel (SDS-PAGE), and transferred to polyvinylidene difluoride (PVDF) membranes. After being blocked with 5 % fat-free milk in TBS-Tween 20, the membranes were incubated with primary antibodies overnight at 4 °C. Then, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies and visualized by chemiluminescence (ECL) plus substrate. Rabbit anti-p-ERK, rabbit anti-ERK, rabbit anti-p-p38 MAPK, rabbit anti-p38 MAPK, rabbit anti-p-JNK, and rabbit anti-JNK antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Rabbit anti-Bcl-2 and mouse anti-GAPDH antibodies were supplied by Santa Cruz Biotechnology (Santa Cruz, CA, USA). The protein levels were represented by the ratio of optical densities in their bands normalized against GAPDH.
U0126 treatment
MEK1/2 inhibitor U0126 (Cell Signaling Technology, Beverly, MA, USA) was dissolved in dimethyl sulfoxide (DMSO). K562/A02 cells were placed to plates for 12 h and incubated with 0, 5, 10, 20 or 40 μM of U0126 for 0, 12, 24 or 48 h, respectively. Control cells received DMSO (solvent) only.
Statistical analysis
All statistical analyses were performed using SPSS software (IBM SPSS version 19.0, USA), and a P < 0.05 two sided was considered significant. Continuous variables were presented as mean ± SD and compared by independent sample t test and one-way ANOVA.
Results
Knockdown of GCS partially reversed cell resistance to ADM through upregulation of apoptosis in K562/A02 cells
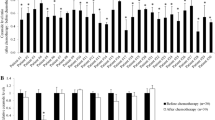
First, we measured the IC50 values of ADM in K562/A02 cells after siRNA treatments. Transfected cells were incubated with ADM for 72 h, followed by CCK-8 assay. As shown in Fig. 1b, blocking GCS expression dramatically downregulated the IC50 value of ADM, while, the negative control siRNA had no obvious effect on the sensitivity of K562/A02.

GCS knockdown increased the cell sensitivity to ADM via apoptosis enhancement in K562/A02 cells. a Cell apoptosis analysis. K562/A02 cells were pretreated with siRNAs and then exposed to 10 μg/mL ADM for 48 h. b IC50 values of ADM in K562/A02 after different siRNAs treatments. c Effect of GCS knockdown on Bcl-2 mRNA expression. d Effect of GCS knockdown on Bcl-2 protein level. Data were recorded as mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, # P < 0.001 compared with the negative control group. In addition, MOCK represented another control in which Lipofectamine™2000 was only used
To investigate the function of GCS in terms of drug-induced apoptosis, cells were treated with 10 μg/mL ADM for 48 h after siRNA transfection. Annexin-V/PI assay was employed and FCM showed that the percentage of ADM-induced apoptosis in K562/A02 cells with GCS suppression was significantly higher than that in cells without GCS suppression (P < 0.01; Fig. 1a).
In addition, the expression of apoptosis-related gene Bcl-2 was examined by real-time PCR and Western blotting analysis. The results showed that knockdown of GCS significantly decreased the mRNA and protein levels of Bcl-2 in K562/A02 (Fig. 1c, d). The inhibition efficiency of siRNA against GCS was confirmed by examining GCS mRNA expression (Fig. 1c).
Suppression of MAPK kinase pathway by GCS knockdown
To reveal the involvement of MAPK kinase pathways in GCS knockdown-induced cell apoptosis, total and phosphorylated ERK, JNK and p38 MAPK were measured by Western blotting. As shown in Fig. 2, GCS knockdown led to remarkable reduction in phosphorylated ERK (P < 0.001), but did not apparently influence total ERK. In addition, knockdown of GCS did not affect other MAPK kinases expressions even their phosphorylated forms.

Effects of GCS knockdown on MAPK kinases expression. Levels of total and phosphorylated MAPK kinases were detected by Western blotting in K562/A02 cells with GCS knockdown. GAPDH was immunodetected as the internal standard. # P < 0.001; n = 3
MEK1/2 inhibitor U0126 sensitized K562/A02 cells to ADM
Next, to evaluate the role of ERK signaling in GCS-mediated drug resistance, U0126 in combination with ADM was used. As expected, p-ERK expression was suppressed by U0126 in a dose- and time-dependent manner (Fig. 3d, f). As shown in Fig. 3a, 10 μg/mL of ADM was largely ineffective in inducing apoptosis. However, 20 μM of U0126, which had slightly toxic effect, was co-administered, and the extent of apoptosis in resistant cells increased significantly. Subsequently, K562/A02 cells were exposed to increasing concentrations of ADM in combination with U0126 or alone for 72 h. CCK-8 assay showed that the IC50 value of the combined ADM and U0126 treatment was lower significantly than that of ADM alone (P < 0.001; Fig. 3b), thereby suggesting that downregulation of p-ERK expression could enhance the efficacy of ADM to destroy the drug-resistant leukemia cells.

U0126 treatment increased the cell sensitivity to ADM via apoptosis enhancement. a Apoptosis of K562/A02 cells exposed to 10 μg/mL ADM in the presence or absence of U0126 (20 μM). Cells were pretreated with U0126 for 1 h and then treated with ADM for 48 h. b U0126 increased the sensitivity of K562/A02 cells to ADM. Cells were treated with increasing ADM alone or combination of ADM with U0126 (20 μM) for 72 h followed by CCK-8 assay. c, d Dose-dependent effect of U0126 on Bcl-2 mRNA (c) and protein (d) levels. Cells were exposed to U0126 for 48 h at indicated concentrations. e, f Time-dependent effect of U0126 on Bcl-2 mRNA (e) and protein (f) levels. K562/A02 cells were exposed to 20 μM U0126 for the indicated times. *P < 0.05, # P < 0.001; n = 3
Downregulation of Bcl-2 by ERK signaling pathway inhibition
Furthermore, we tried to understand whether the expression of Bcl-2 was regulated by ERK activation. The mRNA expression level of Bcl-2 was significantly decreased by U0126 treatment in a dose- and time-dependent manner (Fig, 3c, e). To verify the results observed in real-time PCR, Western blotting was performed to further examine the protein expression of Bcl-2 (Fig. 3d, f). In accordance with previous findings, Bcl-2 protein was also downregulated with U0126 treatment.
Resumption of ceramide after GCS knockdown inhibited ERK signaling and Bcl-2 expression
To further elucidate the dynamics of ceramide metabolism in GCS-induced activation of ERK signaling, endogenous ceramide in K562/A02, treated with siRNAs, in the absence or presence of ADM was measured by LC/MS (Fig. 4a). The intracellular levels of ceramide were increased about (2.10 ± 0.27)-fold in GCS siRNA group. While almost no increase in ceramide levels was observed in cells incubated with ADM alone. Importantly, combination of GCS siRNA with ADM treatment caused a significant enhancement in ceramide levels [(6.83 ± 0.90)-fold], suggesting that knockdown of GCS increased ADM-induced ceramide accumulation in K562/A02 cells.

GCS knockdown induced ERK and Bcl-2 inhibition through ceramide. a Ceramide analysis. K562/A02 cells were transfected with siRNAs (100 nM) and further incubated for 24 h in the absence or presence of 10 μg/mL ADM. Ceramide content was quantified and normalized to inorganic phosphorus (Pi), and was shown as the percentage of control group. b Effects of ceramide exposure on p-ERK and Bcl-2 expression. Cells were treated with C6-ceramide at indicated concentrations for 24 h and the activation of ERK signaling, as well as Bcl-2 expression was evaluated. *P < 0.05, # P < 0.001; n = 4
In agreement with other published reports showing that ceramide inhibited the phosphorylation and activation of ERK in gemcitabine-resistant PANC-1 cells [9], we employed C6-ceramide and found that phosphorylation of ERK, as well as Bcl-2 expression, was decreased upon C6-ceramide exposure (Fig. 4b). Altogether, our findings demonstrated that knockdown of GCS stimulated ceramide-induced ERK inactivation and Bcl-2 inhibition in K562/A02 cells.
Discussion
MDR enables cells to escape from cytotoxicity of chemotherapy and exert drug resistance phenotype, remaining a major cause of leukemia treatment failure. As well as known mechanisms for years, elevated GCS has been reported in drug-resistant cancers and correlated with poor response of cells to anti-cancer agents. The relationship between GCS and drug resistance has been studied in diverse tumors. Besides breast cancer [10, 11], GCS is overexpressed in such as leukemia, melanoma, colon cancer, renal cell cancer, and head and neck epidermoid carcinoma that display resistance to chemotherapy [12–15]. Our previous research also demonstrated that the K562/A02 cell line with higher exhibition of GCS was 115-fold more resistant to ADM and 36-fold more resistant to vincristine compared with its sensitive counterpart K562 [4]. Genetic knockdown or pharmacological inhibition of GCS led to increased cell apoptosis and restored the sensitivity to drugs [12, 16, 17]. For example, GCS-targeted siRNA or GCS-specific inhibitor PPMP sensitized K562/A02 cells to ADM by 2.1- [2] or 4.4-fold [4], respectively. By contrast, upregulation of GCS through introducing GCS into wild-type breast cancer MCF-7 cells enhanced EC50 values of ADM and ceramide by 11- and 5-fold, respectively [18]. Based on that fact, high expression of GCS plays an important role in MDR.
As the adaptive mechanisms emerge following conventional treatment, cancer cells obtain abilities to evade apoptotic execution, contributing to the development of drug resistance [19]. To reveal the relationship between GCS and apoptosis signaling, our previous study demonstrated higher expression of Bcl-2 in K562/A02 cells than that in sensitive K562. However, both of the cell lines shared the same expression level of Bax [5]. Bcl-2 mRNA and protein were downregulated after GCS knockdown, while no difference of Bax was observed. In parallel with the previous data, Annexin-V-FITC/PI staining assay indicated that knockdown of GCS enhanced cellular apoptosis and then sensitized K562/A02 cells to ADM in present study.
Numerous investigations demonstrated that the intracellular signal transduction of apoptosis in cancer cells was closely related to MAPK pathways upon extracellular stimuli. Subsequently, we focused on the role of MAPK pathways in apoptosis caused by GCS inhibition in K562/A02 cells. In addition to enhanced expression of p-ERK in K562/A02 compared with its parental line (K562) [20], p-ERK inhibition, regardless of total ERK unchanged, was detected when GCS was knockdown. Whereas, JNK or p38 MAPK activation did not change appreciably with all treatments, suggesting that ERK signaling instead of JNK or p38 MAPK participated in GCS-induced drug resistance in K562/A02.
To investigate whether ERK activation mediated GCS-induced Bcl-2 enhancement, K562/A02 cells were exposed to increasing concentrations of U0126 for various amounts of time. The treatment of U0126 resulted in a marked reduction in mRNA and protein expressions of Bcl-2 in a dose- and time-dependent manner. These findings were consistent with published reports demonstrating a requirement for ERK activation in sustained expression of Bcl-2 in K562 [20]. Significantly, coadministration of U0126 with a pharmacologically achievable concentration of ADM, which failed to destroy cells, caused a substantial increase in apoptosis and sensitivity to ADM in spite of the slight toxic effect of U0126 on K562/A02 cells. Moreover, activation of ERK pathway has been linked to GCS-mediated P-gp upregulation revealed in our previous study [6, 21], also suggesting a pivotal role of ERK in GCS knockdown-induced reversal of drug resistance. Aside from Bcl-2, several other apoptosis-related proteins, such as Bcl-XL and Mcl-1, had been suppressed when exposed to MEK inhibitors [20, 22]. In this regard, interactions between ERK and Bcl-2 family under the regulation of GCS might be noteworthy. The possibility that the expressions of Bcl-XL and Mcl-1 correlate with GCS in K562/A02 would be of great interest for further exploration.
With regard to inhibition of ERK signaling upon GCS knockdown, which mechanisms or factors primarily involved were further explored in the present study. GCS is a critical enzyme in ceramide glycosylation and catalyzes the conversion of apoptotic ceramide to GlcCer. Blocking ceramide glycosylation by inhibition of GCS results in an increase in ceramide level, inhibiting tumor progression [23] or sensitizing cancer cells to chemotherapy [24]. Mechanistically, ceramide exhibits pro-apoptotic actions through JNK [25, 26] and p38 MAPK [26, 27] phosphorylation, and ERK inhibition [28]. In spite of ERK activation occurring in response to ceramide in some other cell types, the results from LC/MS and Western blotting in the present study indicated that GCS knockdown induced ERK signaling inhibition by ceramide, consistent with the findings of Jiang et al. [9], further demonstrating that low expression level of ceramide, as a cellular messenger, contributed to GCS-induced drug resistance.
In conclusion, GCS positively regulated Bcl-2 expression and cell apoptosis through ERK signaling, conferring chronic leukemia K562/A02 cells drug resistance, as suggested by the present findings. More so, increased metabolism of ceramide by GCS was involved in GCS-induced ERK activation and Bcl-2 overexpression. Knockdown of GCS had a pro-apoptotic effect on drug-resistant cancers building up a strategic treatment for refractory malignancy. However, mechanisms of GCS-mediated MDR are very complex. It is usually a synergistic result of several mechanisms. Reversal of drug resistance should be concentrated on and needs more in vivo experiments to be executed.
References
Liu YY, Yu JY, Yin D, Patwardhan GA, Gupta V, Hirabayashi Y, et al. A role for ceramide in driving cancer cell resistance to doxorubicin. FASEB J. 2008;22:2541–51.
Zhang YY, Xie KM, Yang GQ, Mu HJ, Yin Y, Zhang B, et al. The effect of glucosylceramide synthase on P-glycoprotein function in K562/AO2 leukemia drug-resistance cell line. Int J Hematol. 2011;93:361–7.
Robertson LE, Chubb S, Meyn RE, Story M, Ford R, Hittelman WN, et al. Induction of apoptotic cell death in chronic lymphocytic leukemia by 2-chloro-2′-deoxyadenosine and 9-beta-d-arabinosyl-2-fluoroadenine. Blood. 1993;81:143–50.
Xie P, Shen YF, Shi YP, Ge SM, Gu ZH, Wang J, et al. Overexpression of glucosylceramide synthase in associated with multidrug resistance of leukemia cells. Leuk Res. 2008;32:475–80.
Liu Y, Xie KM, Yang GQ, Bai XM, Shi YP, Mu HJ, et al. GCS induces multidrug resistance by regulating apoptosis-related genes in K562/AO2 cell line. Cancer Chemother Pharmacol. 2010;66:433–9.
Li YL, Xie KM, Zhang YY, Mu HJ, Zhang B, Zou J. et al [Effect of glucosylceramide synthase on P-gp expression by ERK signal transduction pathway in leukemia multi-drug resistance cell line]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2012;29:270–4.
Kim BK, Kim HM, Chung KS, Kim DM, Park SK, Song A, et al. Upregulation of RhoB via c-Jun N-terminal kinase signaling induces apoptosis of the human gastric carcinoma NUGC-3 cells treated with NSC12618. Carcinogenesis. 2011;32:254–61.
Milhas D, Andrieu-Abadie N, Levade T, Benoist H, Segui B. The Tricyclodecan-9-yl-xanthogenate D609 triggers ceramide increase and enhances FasL-induced caspase-dependent and -independent cell death in T Lymphocytes. Int J Mol Sci. 2012;13:8834–52.
Jiang Y, DiVittore NA, Kaiser JM, Shanmugavelandy SS, Fritz JL, Heakal Y, et al. Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer Biol Ther. 2011;12:574–85.
Liu YY, Patwardhan GA, Xie P, Gu X, Giuliano AE, Cabot MC. Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. Int J Oncol. 2011;39:425–31.
Lavie Y, Cao H, Volner A, Lucci A, Han TY, Geffen V, et al. Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. J Biol Chem. 1997;272:1682–7.
Gouaze V, Yu JY, Bleicher RJ, Han TY, Liu YY, Wang H, et al. Overexpression of glucosylceramide synthase and P-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Mol Cancer Ther. 2004;3:633–9.
Ponnusamy S, Meyers-Needham M, Senkal CE, Saddoughi SA, Sentelle D, Selvam SP, et al. Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010;6:1603–24.
Lavie Y, Cao H, Bursten SL, Giuliano AE, Cabot MC. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J Biol Chem. 1996;271:19530–6.
Liu YY, Hill RA, Li YT. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv Cancer Res. 2013;117:59–89.
Gouaze-Andersson V, Yu JY, Kreitenberg AJ, Bielawska A, Giuliano AE, Cabot MC. Ceramide and glucosylceramide upregulate expression of the multidrug resistance gene MDR1 in cancer cells. Biochim Biophys Acta. 2007;1771:1407–17.
Gouaze V, Liu YY, Prickett CS, Yu JY, Giuliano AE, Cabot MC. Glucosylceramide synthase blockade down-regulates P-glycoprotein and resensitizes multidrug-resistant breast cancer cells to anticancer drugs. Cancer Res. 2005;65:3861–7.
Liu YY, Han TY, Giuliano AE, Cabot MC. Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J Biol Chem. 1999;274:1140–6.
Indran IR, Tufo G, Pervaiz S, Brenner C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim Biophys Acta. 2011;1807:735–45.
Yu C, Krystal G, Varticovksi L, McKinstry R, Rahmani M, Dent P, et al. Pharmacologic mitogen-activated protein/extracellular signal-regulated kinase kinase/mitogen-activated protein kinase inhibitors interact synergistically with STI571 to induce apoptosis in Bcr/Abl-expressing human leukemia cells. Cancer Res. 2002;62:188–99.
Zhang X, Xie K, Zou J, Li Y, Mu H, Zhang B. et al [NF-kappa B mediates the effect of glucosylceramide synthase on P-glycoprotein modulation in a drug-resistance leukemia cell line]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2014;31:34–8.
Nguyen TK, Rahmani M, Harada H, Dent P, Grant S. MEK1/2 inhibitors sensitize Bcr/Abl+ human leukemia cells to the dual Abl/Src inhibitor BMS-354/825. Blood. 2007;109:4006–15.
Weiss M, Hettmer S, Smith P, Ladisch S. Inhibition of melanoma tumor growth by a novel inhibitor of glucosylceramide synthase. Cancer Res. 2003;63:3654–8.
Patwardhan GA, Zhang QJ, Yin D, Gupta V, Bao J, Senkal CE, et al. A new mixed-backbone oligonucleotide against glucosylceramide synthase sensitizes multidrug-resistant tumors to apoptosis. PLoS One. 2009;4:e6938.
Nica AF, Tsao CC, Watt JC, Jiffar T, Kurinna S, Jurasz P, et al. Ceramide promotes apoptosis in chronic myelogenous leukemia-derived K562 cells by a mechanism involving caspase-8 and JNK. Cell Cycle. 2008;7:3362–70.
Chen CL, Lin CF, Chang WT, Huang WC, Teng CF, Lin YS. Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood. 2008;111:4365–74.
Wang J, Lv XW, Du YG. Potential mechanisms involved in ceramide-induced apoptosis in human colon cancer HT29 cells. Biomed Environ Sci. 2009;22:76–85.
Bourbon NA, Yun J, Berkey D, Wang Y, Kester M. Inhibitory actions of ceramide upon PKC-epsilon/ERK interactions. Am J Physiol Cell Physiol. 2001;280:C1403–11.
Acknowledgments
This work was supported by a Natural Science Foundation of China (NFSC) Grant (81000527).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Wang, Q., Zou, J., Zhang, X. et al. Glucosylceramide synthase promotes Bcl-2 expression via the ERK signaling pathway in the K562/A02 leukemia drug-resistant cell line. Int J Hematol 100, 559–566 (2014). https://doi.org/10.1007/s12185-014-1679-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-014-1679-7