
Abstract
Purpose of Review
Lipoprotein(a) (Lp(a)) is a proinflammatory and atherogenic molecule that is emerging as an important biomarker of cardiovascular (CV) risk. It has been implicated in the pathogenesis of both atherosclerotic cardiovascular disease (ASCVD) and aortic valve stenosis. An estimated 10–30% of the global population has elevated Lp(a) levels. Screening for and treating elevated Lp(a) represents an opportunity to reduce risk for adverse CV events.
Recent Findings
Current guidelines from the American College of Cardiology and National Lipid Association recommend Lp(a) testing in high-risk individuals. The European Atherosclerosis Society takes a stronger stance, recommending once in a lifetime lipoprotein(a) testing across the general population. Few recommendations currently exist regarding treatment of elevated lipoprotein(a).
Summary
Because elevated Lp(a) is an independent risk factor for cardiovascular disease, further screening and treatment is necessary. Ongoing clinical trials with RNA based therapeutics such as antisense oligonucleotides and small interfering RNA show great promise for lowering Lp(a). More data is needed to demonstrate improved cardiovascular outcomes from Lp(a) reduction. Lp(a) should be incorporated into clinical practice as a useful biomarker to guide decisions about ASCVD risk and treatment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lipoprotein A [Lp(a)] is an apolipoprotein B100 (apoB) molecule covalently bonded to glycoprotein apolipoprotein (a) [apo(a)] [1]. Several studies suggest that high Lp(a) levels promote atherosclerosis due to Lp(a)-derived cholesterol entrapment in the intima, inflammatory cell recruitment, and the binding of proinflammatory oxidized phospholipids [1,2,3,4]. Lp(a) is also hypothesized to have a prothrombotic effect which may be due to the structural similarity of apo(a) to plasminogen, the precursor of the fibrinolytic enzyme plasmin [5]. The prothrombotic and atherosclerotic nature of Lp(a) may explain why high Lp(a) levels are consistently associated with manifestations of ASCVD including myocardial infarctions and stroke [1, 6, 7].
Over the last decade, evidence suggests that Lp(a) is the main lipoprotein carrier of phosphocholine containing oxidized phospholipids (OxPL) [8,9,10,11]. OxPL has been recognized as a danger associated molecular pattern (DAMP) by pattern recognition receptors (PRRs) on innate immune cells, leading to pro-inflammatory and plaque destabilizing processes [8, 12,13,14]. This has led to the hypothesis that a major component of risk associated with Lp(a) may be from its OxPL content. Epidemiological studies further demonstrate that higher levels of OxPL on apoB (OxPL-apoB), especially present on Lp(a), can be used as indicators of future ASCVD risk [8, 15, 16]. Over 40 publications corroborate this correlation, with elevated OxPL-apoB levels associated with greater death, MI, and stroke [17,18,19]. Therefore, OxPL-apoB and Lp(a) levels both represent useful markers for assessing ASCVD risk.
Lp(a) levels are found to be highly heritable and thus largely controlled by genetic variants at the LPA ([lipoprotein, Lp(a) gene, OMIM + 152200] locus [1, 20]. One copy variation in Lp(a) which consists of a variable number of kringle (K) IV type 2 repeats (KIV-2) in the LPA gene has been found to determine Lp(a) levels [1]. Specifically, the genetically determined KIV-2 repeat size contributes to 30–70% of the variation in Lp(a) levels and affects the final size of apo(a); larger isoforms are more conductive to protein folding, transport, and secretion [1, 20, 21]. Studies find that a low number of LPA KIV-2 repeats are associated with increased Lp(a) secretion and can increase ASCVD risk [22].
Currently, Lp(a) is most commonly measured via immunoassays. Most use a variety of polyclonal antibodies against apo(a), and react with the highly polymorphic kringle IV type 2 domain [23]. Depending on size of apo(a) and assay calibration, Lp(a) level may be under or overestimated. Lp(a) particles are extremely variable in apo(a) size, lipid content, and glycosylation modification [24]. It may be expressed as molar concentrations (nmol/L) or as mass concentrations (mg/dL). In 2019, the HEART UK consensus statement suggested that Lp(a) concentrations should be expressed in nmol/L [25] and the concentration of Lp(a) should be measured using appropriate antibodies to minimize the effects of isoform size. Although measurement in nmol/L is recommended, data has often been historically collected as mg/dL, so many guidelines stratify risk based on mass concentrations [25]. As with other measurements, variability between labs, calibration, and reporting can make clinical interpretation of data challenging. Current techniques to measure LDL-C include Lp(a), and the reported LDL-C is actually a combination of LDL-c and Lp(a) [26•]. In patients with elevated Lp(a), Lp(a) may constitute a significant portion of reported LDL-c. Therefore, interpretation of LDL-c must be corrected for by subtracting Lp(a), noted as either LDL-Ccorr = laboratory LDL-C − directly measured Lp(a)-C or by the Dahlén formula, as LDL-CcorrDahlén = laboratory LDL-C − [Lp(a) mass × 0.30] [26•]. For example, in a patient with an Lp(a) of 50 mg/dL and an LDL-C of 150, the LDL-Ccorr would be 100, a significantly different value than prior to correction. In addition, when patients do not respond appropriately to statin therapy or have “statin resistance,” Lp(a) should be assessed as this may be a significant contributor to LDL-C.
Optimal assays analyze Lp(a) without confounding from apo(a) heterogeneity and with correction of LDL-c, noted as LDL-Ccorr. Widescale implementation of immunoassays that minimize sensitivity to apolipoprotein(a) isoform size and account for LDL-c separate from Lp(a) are the next steps in clinically applicable screening.
Prevalence
According to the National Lung and Blood report published in 2018, an estimated 1.43 billion people globally have Lp(a) levels greater than 50 mg/dl with a prevalence of 10–30% [27]. The population mean and median levels of Lp(a) are found to vary by ethnicity [27]. Blacks, both Africans and African Americans, have 2 to 3 times higher levels of Lp(a) when compared to their white counterparts. Latin Americans have lower levels than whites and Chinese individuals have lower levels than those of Indian origin [27,28,29,30]. While different ethnic populations have differing prevalence of elevated Lp(a), the risk attributed to higher Lp(a) levels varies [28, 30].
For certain ethnic populations such as South Asians, lipoprotein(a) may account for cardiovascular disease burden that is not yet fully addressed. Current guidelines recommend reduction of LDL-c to minimize ASCVD risk. However, the IMPROVE-IT trial demonstrated that aggressive LDL therapy with statins and ezetimibe did not fully eliminate CVD risk [31]. The INTERHEART study demonstrated the highest odds ratio (OR) for acute myocardial infarction from high Lp(a) concentrations in South Asians (OR 2.14), followed by South East Asians (OR 1.83), Latin Americans (OR 1.67), Chinese (OR 1.62), and Europeans (OR 1.36) [27]. These higher risk populations could benefit from more rigorous Lp(a) treatment cutoffs. The opportunity to screen and subsequently treat elevated lipoprotein(a) remains unexplored territory and presents a further opportunity to address ASCVD burden more thoroughly. Thus, further discussion of treatment guidelines should take into consideration a multimodal and personalized approach to lipid testing and treatment that includes Lp(a). The discussion could further decrease discrepancies in cardiovascular disease burden for ethnic populations with elevated Lp(a) levels (Fig. 1).

Estimated prevalence of elevated lipoprotein(a) globally
Lp(a) and Association with ASCVD and Aortic Stenosis
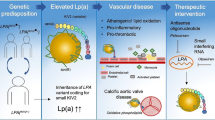
Multiple studies during the last decade have implicated Lp(a) as a risk factor for both ASCVD and calcific aortic valve disease [32]. A curvilinear relationship has been shown between Lp(a) levels and ASCVD risk, with lower risk at levels < 50 mg/dL or < 75 nmol/L. Increased risk has been seen linearly with higher levels [6, 33, 34]. The desirable and optimal test result range of Lp(a) is < 14 mg/dL. The highest risk range is > 50 mg/dL. Studies have demonstrated that risk is equally distributed between men and women.
The linkage between Lp(a) and atherogenesis may rely on the highly oxidative property of Lp(a) [35]. Compared to LDL, Lp(a) is more likely to undergo oxidation, facilitating higher uptake by macrophages before transforming into foam cells, the primary precursors of atherosclerosis [35]. Higher plasma levels of Lp(a) (> 50 mg/dL) have also been found to induce endothelial dysfunction, contributing to its pro-atherogenic role [35, 36].
Findings from several epidemiological, genome-wide association, and Mendelian randomization studies suggest that Lp(a) is also a major risk factor for aortic valve calcification and aortic stenosis [17]. Specifically, aortic valve leaflets contain oxidized lipids, apoB, apo(a), macrophages, and T-cells [37,38,39]. When aortic valves are subjected to mechanical stress, apo(a) binds to fibrin on injured endothelium, allowing Lp(a) to enter into the valve. OxPL, a pro-inflammatory molecule present on Lp(a), promotes calcification of vascular cells. The subsequent complex of Lp(a)-OxPL induces pro-inflammatory and pro-calcific properties on the valve, accelerating progression of aortic stenosis.
Screening for Lipoprotein(a)
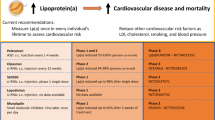
Expert opinion varies on when to screen for elevated Lp(a). The American College of Cardiology, American Heart Association, and National Lipid Association (ACC/AHA/NLA) Task Force established Clinical Practice Guidelines on the Management of Blood Cholesterol in 2018, which recommends testing for individuals at high risk for ASCVD [40,41,42]. The National Lipid Association offers a more detailed view on the specific populations in which Lp(a) screening might be warranted. The NLA recommends that Lp(a) screening should be considered when there is a family history of premature ASCVD, personal history of ASCVD not explained by other major risk factors [41], and primary severe hyperlipidemia. It further recommends Lp(a) testing to support the use of statins in those who are in the borderline ASCVD risk category [43]. However, the ACC/AHA/NLA do not recommend routine screening in low-risk patients.
Meanwhile, the European Atherosclerosis Society recommend testing in patients with a personal history of premature cardiovascular disease, recurrent cardiovascular disease while on statin therapy, ≥ 10% 10-year risk of cardiovascular disease, family history of elevated Lp(a)/premature cardiovascular disease, and familial hypercholesterolemia. The European Society of Cardiology has the strongest guideline recommendation and encourages testing for Lp(a) at least once in a person’s lifetime [44]. There is currently no recommendation regarding testing Lp(a) in pediatric patient populations.
Treatment of Elevated Lp(a)
Reduction of LDL-c remains the cornerstone for ASCVD management and for minimizing cardiovascular events [32, 45]. However, studies have shown that even in patients with LDL levels at goal, Lp(a) may account for residual cardiovascular risk [46,47,48]. Since Lp(a) contributes to atherosclerosis and remains an independent risk factor for cardiovascular events, there is an unmet need for specific Lp(a)-lowering therapies.
Historically, niacin (i.e., vitamin B3) was the only approved lipid-lowering drug that showed a potent reduction in Lp(a) levels [44]. Niacin lowers Lp(a) by silencing apo(a) gene expression in hepatocytes [49]. A dose-dependent decrease in Lp(a) levels of 25% and 38% with 2 and 4 g of daily niacin has been reported [50, 51]. However, despite reduction in Lp(a) levels, the effect of niacin has not demonstrated any overall reduction of CV events [52, 53]. Furthermore, a randomized double blind trial found no difference in CV events between simvastatin + placebo and simvastatin + niacin [54]. Therefore, niacin has not been shown to improve morbidity and mortality from ASCVD beyond statin therapy. Given the high side-effect profile of niacin at effective doses and lackluster data regarding its benefits, niacin is not widely used clinically.
In the FOURIER and ODYSSEY Outcome trials, PCSK9 inhibitors reduce Lp(a) levels by 25–30% [55, 56•, 57]. Unlike niacin, the use of PCSK9 inhibitors led to improved cardiovascular outcomes [58, 59]. A post hoc analysis of the FOURIER trial revealed an overall 16% reduction in major adverse cardiovascular events in patients treated with evolocumab, independent of LDL cholesterol reduction [59]. Another analysis of FOURIER revealed that treatment with evalocumab also lowered the progression of aortic valve stenosis and the subsequent need for aortic valve replacement [60]. The ODYSSEY Outcome trial showed that changes in Lp(a) levels after alirocumab therapy reduced the risk of major adverse cardiovascular events by 0.6% for each 1 mg/dL reduction in Lp(a) levels, independent of LDL cholesterol reduction [61]. These results indicate clinically meaningful benefits from PCSK9 therapy for patients with higher baseline Lp(a) levels. Although treatment of elevated Lp(a) with PCSK9 inhibitors has not yet received any strong guideline recommendation, these drugs appear to be promising candidates for the treatment of the ‘residual’ risk carried by high Lp(a) levels [44]. Encouragingly, and in contrast to niacin, PCSK9 inhibitors are well tolerated. However current evidence for benefits of PCSK9 inhibitors has only come from secondary sub analyses.
Statins are a mainstay of therapy in primary and secondary prevention of cardiovascular (CV) disease. They effectively reduce LDL cholesterol and significantly impact CV mortality [44]. Effects of statins on the metabolism of Lp(a) is not fully understood. Sub- and meta-analyses of statin trials have hinted at potential benefits of statin therapy for Lp(a) levels, but this observation was not replicated in further studies [17, 47, 62, 63]. One meta-analysis of six randomized controlled trials even demonstrated a significant increase in Lp(a) levels following statin therapy [64]. The role of statins on Lp(a) levels remains a mixed picture. Ezetimibe, a weak LDL-lowering agent, has shown to reduce Lp(a) levels by 7% [65].
A more drastic approach to treating elevated Lp(a) is lipoprotein apheresis. Although an invasive procedure, lipoprotein apheresis is generally safe with most complications related to the puncture site [66]. During apheresis, plasma is first separated from the blood and lipids are then selectively absorbed and removed from the plasma. The best evidence for potential benefits of Lp(a) lowering has come from studies where patients received lipid apheresis for Lp(a) > 60 mg/dL and recurrent ASCVD events despite optimal LDL-C reduction and other secondary prevention measures. The Pro(a)LiFe study (Prospective Documentation of Isolated Lp(a)-Elevation With Progressive Cardiovascular Disease and Lipoprotein Apheresis for Effective Treatment of Hyperlipoproteinemia) enrolled 170 patients and demonstrated that once patients receive apheresis, the event rate for ASCVD events decreases by 70 to 80% compared with a pre-apheresis event rate [67]. Apheresis has also been shown to improve coronary blood flow and reduce angina frequency in patients with refractory angina and Lp(a) > 50 mg/dL, though this data comes from a small sample size of 20 patients [68]. Apheresis can lower Lp(a) acutely by 70%, but because of rapid production by the liver, levels generally normalize within a week, with a net time-averaged reduction of only 35 to 40%. This effect is only slightly higher than treatment with PCSK9 inhibitors [69] The MultiSELECt trial (Effect of Lp(a) Elimination by Lipoprotein Apheresis on Cardiovascular Outcomes) addresses the question of whether patients undergoing apheresis have improved outcomes compared to standard of care, and results are pending. Apheresis has multiple effects on all apo B-containing lipoproteins that together complicate whether the apheresis process produces genuine long term benefit.
Hormonal Regulation
The metabolism and regulatory mechanisms of Lp(a) are not well elucidated. It is known that there is a relationship between Lp(a) and various hormones, including estrogen, testosterone, growth hormone, and thyroid hormone, and that the biomarker is hormonally regulated [70]. For decades, studies have shown that estrogen and testosterone can lower Lp(a) levels [71]. Estrogen supplementation in postmenopausal women has been associated with decreased Lp(a) levels [70, 71]. Sub-analysis of the Heart and Estrogen/Progestin Replacement Study (HERS) showed a 15 mg/dL mean reduction in Lp(a) levels with estrogen and progestin therapy [72]. The mechanism by which estrogen decreases Lp(a) is thought to be due to the hormone’s activity on the Apo(a) promoter [73]. Studies have additionally demonstrated that a history of premature menopause is a risk factor for elevated Lp(a) [70]. Estrogen is currently not recommended as a treatment modality for elevated Lp(a) given the complex regulation of multiple lipid molecules such as apolipoprotein A1, HDL cholesterol, triglyceride levels. Like estrogen, selective estrogen receptor modulators such as raloxifene have been shown to lower serum Lp(a) [74]. For male patients, low testosterone has been associated with elevated Lp(a) [75]. One study showed that 17.1% of men with low testosterone had Lp(a) over 3 × the upper limit of normal compared to 8.1% of men with normal testosterone. Subsequent testosterone treatment reduced Lp(a) levels significantly [75]. However, given that both estrogen and testosterone can have adverse cardiovascular effects, it is not recommended that patients be given supplementation with estrogen or testosterone for the sole purpose of reducing Lp(a) levels.
In addition to regulation by sex hormones such as estrogen and testosterone, Lp(a) is affected by growth and thyroid hormone. While growth hormone supplementation is associated with decreased LDL and total cholesterol, it has been shown to increase Lp(a) in a dose dependent fashion [76]. This is hypothesized to be due to growth hormone’s effect on the synthesis and secretion of apo(a) as well as interaction with hepatic LDL receptors. Mouse models have further shown that the effect of growth hormone on Lp(a) works independently of IGF-1 [77]. It has also been well documented that the hypothyroid state is associated with lipid abnormalities, and Lp(a) is no exception [78]. Hypothyroidism correlates with elevated Lp(a), thought to be due to severely decreased catabolism during the hypothyroid state [79].
While we know that a relationship between these hormones and Lp(a) exists, there is little data to support how to use this understanding to target further reduction in Lp(a). Further consideration should also be given regarding how to best identify and treat populations with hormonal abnormalities affecting Lp(a) levels. To reach a definitive conclusion on the role of testosterone replacement therapy in the context of preventive cardiology, more research is needed, as the cumulative evidence is currently unclear. L-carnitine may also lower Lp(a) levels but has been linked to elevated trimethylamine N-oxide (TMAO), potentially leading to atherosclerosis [80]. More therapies continue to arise, and further studies are needed to analyze if cardiac outcomes are improved by hormonal therapy.
Future Directions
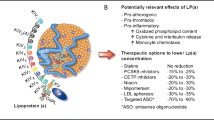
In clinical practice, no clear studies have been published regarding the utility of aspirin in populations with elevated Lp(a). Lp(a) can be atherothrombotic through several mechanisms: atherogenic via the LDL-like moiety, proinflammatory via OxPL content, and potentially antifibrinolytic via its apo(a) moiety, tilting the balance toward thrombosis [23]. Given the thrombogenic nature of Lp(a), low-dose aspirin as a treatment intervention in elevated Lp(a) could be considered [81, 82]. Although recent trials on aspirin (i.e. ASPREE, ASCEND) did not confirm any overall beneficial role of aspirin in primary prevention, there may be a role for the drug in patients with elevated Lp(a) [83, 84]. Further areas of study could demonstrate whether patients with elevated Lp(a) may benefit from antiplatelet therapy with potential reduction in major adverse cardiovascular events. Additionally, new treatments targeting Lp(a) are emerging such as PCSK9 inhibitors, antinuclear sense RNA, and small interfering RNA. Investigation of their benefit on cardiovascular morbidity and mortality are currently being explored.
RNA Therapeutics for Lp(a)
Antisense Oligonucleotides
Because Lp(a) has no enzymatic activity and high plasma levels in individuals who are at the greatest risk, it cannot be easily targeted with small molecules or monoclonal antibodies [85]. RNA therapeutics, specifically antisense oligonucleotides (ASOs), represent an elegant method to reduce circulating Lp(a). Apo(a) is primarily (> 99%) synthesized by hepatocytes and is then assembled into an Lp(a) particle, either intracellularly on the extracellular surface of hepatocytes or in the circulation by binding to an LDL-like particle [85,86,87,88].
Small amounts of apo(a) reside on VLDL particles, and kinetic studies do not suggest that VLDL is a significant precursor to Lp(a). There are no kinetic studies that elucidate the precise pathways, which have thus far been targeted with apo(a)-lowering treatments. ASOs injected subcutaneously bind to plasma proteins and then enter the hepatocyte nucleus and bind to apo(a) mRNA. The formation of an ASO/mRNA duplex activates RNAse H1 to cleave the sense strand and prevent protein translation. Thus, hepatocyte production of apo(a) is significantly inhibited, and Lp(a) levels are reduced. The liver continues to assemble and secrete VLDL and other lipoproteins.
Potential of ASOs in lowering Lp(a) was first documented in Lp(a) transgenic mice, in which treatment with mipomersen reduced Lp(a) by 75% [86]. Potent reduction of hepatic Apo B-100 production halted serum apo B-100 production and Lp(a) particle assembly. In a second study with Lp(a) transgenic mice, an apo(a)-specific ASO, 144,367 reduced Lp(a) levels by 86% and paved the way for the development of optimized ASOs for human applications. In healthy human subjects, mipomersen, an ApoB-100 ASO, lowered Lp(a) by an average of 21% [89]. This model demonstrates proof of concept that an ASO based drug platform could lower Lp(a). However, mipomersen is not used clinically for elevated Lp(a) due to severe adverse side effects including significant liver toxicity; the drug is also not FDA approved for reducing Lp(a).
These novel approaches are currently being investigated in multiple clinical trials [90, 91]. Currently a phase 3 trial, the HORIZON trial, is examining the effect of pelacarsen [92]. Pelacarsen aims to reduce apo(a) production by the liver, and therefore Lp(a) using an ASO directed against mRNA of apo(a) [93]. Results from phase 2 trials demonstrated a dose dependent reduction in Lp(a) levels with mean reduction of 80% at the highest dose [94]. The drug is injected as 80 mg monthly, and main side effects seen from the drug were local reactions at the injection site. The data also shows 70–88% decreased OxPL-apoB and OxPL-apo(a), highlighting decreased thrombosis and further suppressed inflammation from reduction of Lp(a). The effectiveness of the drug and favorable safety profile represent promise in decreasing Lp(a) levels. The ongoing HORIZON trial will answer the question if lowering Lp(a) levels will translate into improved cardiovascular outcomes [94].
Small Interfering RNA
Another promising category of drugs entering phase 3 trials are small interfering RNA molecule (siRNA). Generally, siRNA works by inhibiting translation of genes that subsequently prevents protein production. Recently, the first in class siRNA compound inclisiran was FDA approved for lowering LDL. Inclisiran inhibits hepatic synthesis of PCSK9 and lowers Lp(a) by 25%. Cardiovascular outcome trials with inclisiran are pending. The main advantages of inclisiran are its long half-life, and patients receive the drug once every 6 months subcutaneously (after the initial loading dose).
Olpasiran is another siRNA molecule that disrupts translation of messenger RNA (mRNA) that is responsible for Lp(a) particle production [95]. In transgenic mice, Olpasiran reduced Lp(a) levels in a dose dependent manner, where a single dose reduced Lp(a) levels by over 80% from baseline on average [95, 96]. The effects of Olpasiran last for several months, with sustained reduction in Lp(a) levels. The drug is very well tolerated, and human trials with 64 participants have demonstrated minimal adverse effects. The most common treatment emergent adverse effects were headache (10% Olpasiran; 25% placebo) and upper respiratory tract infection (15% Olpasiran; 13% placebo) [97] Other benefits of Olpasiran include its long half-life such that a single dose maintains the lipid lowering effect of the drug for 3–6 months.
SLN360 which is also an siRNA molecule that targets Lp(a) has promising data [98, 99••]. In the phase I APOLLO trial that enrolled 32 participants with Lp(a) > 150 nmol/L, SLN360 demonstrated overall Lp(a) reduction by 96–98% after a single dose of 300 or 600 mg and a reduction of 71% and 81%, respectively, at 5 months compared to baseline [99••]. High doses of the drug also lowered LDL cholesterol by roughly 25%. Minimal adverse effects were noted after administration of the drug, and further research is underway to study if this Lp(a) reduction will translate into clinical benefit of the drug.
The FDA has yet to approve any specific therapy for lowering Lp(a) due to the need for further data regarding improved cardiovascular outcomes from pharmacologically lowering Lp(a). The HORIZON trial is actively pursuing data regarding these outcomes. Drugs such as Pelacarsen, an ASO against Apo(a), Olpasiran and SLN360, siRNA targeting LPA gene, showcase immense potential. Continuing to explore effects of potent therapies to lower Lp(a) with minimal side effects and improved cardiovascular outcomes is a next step in advancing lipid screening and treatment as well as delving more deeply into personalized medicine (Fig. 2).

Lipoprotein(a), association with aortic stenosis, and therapeutic treatments for Lp(a) reduction
Conclusion
Lipoprotein(a) represents a novel biomarker with great potential that can help identify high-risk individuals and guide treatment strategies. The correlation between elevated Lp(a) and coronary artery disease as well as aortic stenosis highlights this biomarker as an important clinical risk factor in cardiovascular disease. Due to its atherogenicity and widespread prevalence, adaptation of robust screening and treatment guidelines represent a future direction for cardiovascular disease prevention and treatment. With ethnic disparities in Lp(a) levels, providers and future guidelines may consider a lower threshold for treatment in higher risk groups. Data thus far has not shown mortality and morbidity benefit from screening and treatment, but further research may better highlight the significant reduction in major adverse cardiovascular events. Due to the atherogenic and proinflammatory nature of Lp(a), aspirin may be potentially utilized for prevention but there is no clear data on this. At the forefront of future Lp(a) treatment is ongoing research into antisense oligonucleotides and small interfering RNA, specifically inhibitors of apo(a) and LPA gene translation. We hope that in the future, lipoprotein(a) becomes incorporated into clinical practice as a useful biomarker that will guide decisions about ASCVD risk and treatment.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Sticchi E, et al. Apolipoprotein(a) Kringle-IV type 2 copy number variation is associated with venous thromboembolism. PLoS ONE. 2016;11:e0149427.
Nielsen LB. Atherogenecity of lipoprotein(a) and oxidized low density lipoprotein: insight from in vivo studies of arterial wall influx, degradation and efflux. Atherosclerosis. 1999;143:229–43.
Sotiriou SN, et al. Lipoprotein(a) in atherosclerotic plaques recruits inflammatory cells through interaction with Mac-1 integrin. FASEB J. 2006;20:559–61.
Nordestgaard BG, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–53.
Anglés-Cano E. Structural basis for the pathophysiology of lipoprotein(a) in the athero-thrombotic process. Braz J Med Biol Res. 1997;30:1271–80.
Collaboration ERF, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–23.
Momiyama Y, et al. Associations between serum lipoprotein(a) levels and the severity of coronary and aortic atherosclerosis. Atherosclerosis. 2012;222:241–4.
van der Valk FM, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134:611–24.
Tsimikas S, et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353:46–57.
Bergmark C, et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–9.
Leibundgut G, et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J Lipid Res. 2013;54:2815–30.
Lee S, et al. Role of phospholipid oxidation products in atherosclerosis. Circ Res. 2012;111:778–99.
Miller YI, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–48.
Leibundgut G, Witztum JL, Tsimikas S. Oxidation-specific epitopes and immunological responses: Translational biotheranostic implications for atherosclerosis. Curr Opin Pharmacol. 2013;13:168–79.
Taleb A, Witztum JL, Tsimikas S. Oxidized phospholipids on apoB-100-containing lipoproteins: a biomarker predicting cardiovascular disease and cardiovascular events. Biomark Med. 2011;5:673–94.
Tsimikas S, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. 2012;60:2218–29.
Yeang C, Wilkinson MJ, Tsimikas S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr Opin Cardiol. 2016;31:440–50.
Tsimikas S, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56:946–55.
Kiechl S, et al. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler Thromb Vasc Biol. 2007;27:1788–95.
Kronenberg F, Utermann G. Lipoprotein(a): resurrected by genetics. J Intern Med. 2013;273:6–30.
Boerwinkle E, Menzel HJ, Kraft HG, Utermann G. Genetics of the quantitative Lp(a) lipoprotein trait III Contribution of Lp(a) glycoprotein phenotypes to normal lipid variation. Hum Genet. 1989;82:73–8.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–9.
Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. 2016;57:1339–59.
Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57:526–37.
Cegla J, et al. HEART UK consensus statement on Lipoprotein(a): a call to action. Atherosclerosis. 2019;291:62–70.
Yeang C, et al. Effect of pelacarsen on lipoprotein(a) cholesterol and corrected low-density lipoprotein cholesterol. J Am Coll Cardiol. 2022;79:1035–1046. Yeang et al. assessed the effect of Pelacarsen on directly measured Lp(a) and LDL-C as a therapeutic option for elevated Lp(a) levels. The study demonstrated that Pelacarsen led to dose-dependent reduction in Lp(a) levels and mild reductions in LDL-c for patients with elevated Lp(a). The highest dose caused a 67% decrease in Lp(a) levels with the effect lasting for months, and the drug was overall well tolerated by study participants.
Enas EA, Varkey B, Dharmarajan TS, Pare G, Bahl VK. Lipoprotein(a): An independent, genetic, and causal factor for cardiovascular disease and acute myocardial infarction. Indian Heart J. 2019;71:99–112.
Anand SS, et al. Elevated lipoprotein(a) levels in South Asians in North America. Metabolism. 1998;47:182–4.
Tavridou A, Unwin N, Bhopal R, Laker MF. Predictors of lipoprotein(a) levels in a European and South Asian population in the Newcastle Heart Project. Eur J Clin Invest. 2003;33:686–92.
Tsimikas S, et al. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71:177–92.
Murphy SA, et al. Reduction in total cardiovascular events with ezetimibe/simvastatin post-acute coronary syndrome: the IMPROVE-IT Trial. J Am Coll Cardiol. 2016;67:353–61.
Thanassoulis G. Lipoprotein (a) in calcific aortic valve disease: from genomics to novel drug target for aortic stenosis. J Lipid Res. 2016;57:917–24.
Clarke R, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–28.
Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117:176–84.
Maranhão RC, Carvalho PO, Strunz CC, Pileggi F. Lipoprotein (a): structure, pathophysiology and clinical implications. Arq Bras Cardiol. 2014;103:76–84.
Wu HD, et al. High lipoprotein(a) levels and small apolipoprotein(a) sizes are associated with endothelial dysfunction in a multiethnic cohort. J Am Coll Cardiol. 2004;43:1828–33.
Dweck MR, Boon NA, Newby DE. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol. 2012;60:1854–63.
Olsson M, Thyberg J, Nilsson J. Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves. Arterioscler Thromb Vasc Biol. 1999;19:1218–22.
O’Brien KD, et al. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler Thromb Vasc Biol. 1996;16:523–32.
Mach F, Baigent C, Catapano AL. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk The Task Force for the management of …. Eur Heart J. 2020.
Grundy SM, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139:e1082–143.
Wilson PWF, et al. Lipid measurements in the management of cardiovascular diseases: Practical recommendations a scientific statement from the national lipid association writing group. J Clin Lipidol. 2021;15:629–48.
Wilson DP, et al. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13:374–92.
Mach F, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–88.
Boffa MB, Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57:745–57.
Borén J, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41:2313–30.
Khera AV, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129:635–42.
Langsted A, Kamstrup PR, Nordestgaard BG. High lipoprotein(a) and high risk of mortality. Eur Heart J. 2019;40:2760–70.
Chennamsetty I, et al. Nicotinic acid inhibits hepatic APOA gene expression: studies in humans and in transgenic mice. J Lipid Res. 2012;53:2405–12.
Carlson LA, Hamsten A, Asplund A. Pronounced lowering of serum levels of lipoprotein Lp(a) in hyperlipidaemic subjects treated with nicotinic acid. J Intern Med. 1989;226:271–6.
Sahebkar A, Reiner Ž, Simental-Mendía LE, Ferretti G, Cicero AFG. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism. 2016;65:1664–78.
Parish S, et al. Impact of apolipoprotein(a) isoform size on lipoprotein(a) lowering in the HPS2-THRIVE study. Circ Genom Precis Med. 2018;11:e001696.
Albers JJ, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J Am Coll Cardiol. 2013;62:1575–9.
Investigators TA-H & The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267.
Cao Y-X, Liu H-H, Li S, Li J-J. A Meta-analysis of the effect of PCSK9-monoclonal antibodies on circulating lipoprotein (a) levels. Am J Cardiovasc Drugs. 2019;19:87–97.
Gaudet D, et al. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol. 2014;114:711–715. Gaudet et al. analyzed data from the ODYSSEY OUTCOMES trial with regards to Lp(a) levels. For patients with hypercholesterolemia, treatment with a PCSK9 inhibitor reduced Lp(a) levels by 30% compared to placebo. Reduction in Lp(a) was only weakly correlated with magnitude of reduction in LDL-C.
Raal FJ, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol. 2014;63:1278–88.
Sabatine MS, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–22.
O’Donoghue ML, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139:1483–92.
Bergmark BA, et al. An exploratory analysis of proprotein convertase subtilisin/kexin type 9 inhibition and aortic stenosis in the FOURIER Trial. JAMA Cardiol. 2020;5:709–13.
Bittner VA, et al. Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J Am Coll Cardiol. 2020;75:133–44.
Takagi H, Umemoto T. Atorvastatin decreases lipoprotein(a): a meta-analysis of randomized trials. Int J Cardiol. 2012;154:183–6.
Van Wissen S, et al. Long term statin treatment reduces lipoprotein (a) concentrations in heterozygous familial hypercholesterolaemia. Heart. 2003;89:893–6.
Tsimikas S, Gordts PLSM, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2020;41:2275–84.
Awad K, et al. Effect of ezetimibe monotherapy on plasma lipoprotein(a) concentrations in patients with primary hypercholesterolemia: a systematic review and meta-analysis of randomized controlled trials. Drugs. 2018;78:453–62.
Schettler VJJ, et al. The German Lipoprotein Apheresis Registry (GLAR) - almost 5 years on. Clin Res Cardiol Suppl. 2017;12:44–9.
Roeseler E, et al. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36:2019–27.
Khan TZ, et al. Apheresis as novel treatment for refractory angina with raised lipoprotein(a): a randomized controlled cross-over trial. Eur Heart J. 2017;38:1561–9.
Moriarty PM, Hemphill L. Lipoprotein apheresis. Cardiol Clin. 2015;33:197–208.
Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), hormone replacement therapy, and risk of future cardiovascular events. J Am Coll Cardiol. 2008;52:124–31.
Kim CJ, Jang HC, Cho DH, Min YK. Effects of hormone replacement therapy on lipoprotein(a) and lipids in postmenopausal women. Arterioscler Thromb. 1994;14:275–81.
Shlipak MG, et al. Estrogen and progestin, lipoprotein(a), and the risk of recurrent coronary heart disease events after menopause. JAMA. 2000;283:1845–52.
Wu JH, Lee IN. Studies of apolipoprotein (a) promoter from subjects with different plasma lipoprotein (a) concentrations. Clin Biochem. 2003;36:241–6.
Ferretti G, et al. Raloxifene lowers plasma lipoprotein(a) concentrations: a systematic review and meta-analysis of randomized placebo-controlled trials. Cardiovasc Drugs Ther. 2017;31:197–208.
Kaplan SA, Lin J, Johnson-Levonas AO, Shah AK, Meehan AG. Increased occurrence of marked elevations of lipoprotein(a) in ageing, hypercholesterolaemic men with low testosterone. Aging Male. 2010;13:40–3.
O’Halloran DJ, Wieringa G, Tsatsoulis A, Shalet SM. Increased serum lipoprotein(a) concentrations after growth hormone (GH) treatment in patients with isolated GH deficiency. Ann Clin Biochem. 1996;33:330–4.
Olivecrona H, et al. Hormonal regulation of serum lipoprotein(a) levels. Contrasting effects of growth hormone and insulin-like growth factor-I. Arterioscler Thromb Vasc Biol. 1995;15:847–9.
Kaliaperumal R, William E, Selvam T, Krishnan SM. Relationship between lipoprotein(a) and thyroid hormones in hypothyroid patients. J Clin Diagn Res. 2014;8:37–9.
Sokolov EI, Metel’skaia VA, Perova NV, Shchukina GN. [Hormonal regulation of lipoprotein metabolism: the role in pathogenesis of coronary heart disease]. Kardiologiia 2006;46:4–9.
Serban M-C, et al. Impact of L-carnitine on plasma lipoprotein(a) concentrations: a systematic review and meta-analysis of randomized controlled trials. Sci Rep. 2016;6:1–11.
Chasman DI, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009;203:371–6.
Shiffman D, et al. Coronary heart disease risk, aspirin use, and apolipoprotein(a) 4399Met allele in the Atherosclerosis Risk in Communities (ARIC) study. Thromb Haemost. 2009;102:179–80.
McNeil JJ, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379:1519–28.
ASCEND Study Collaborative Group et al. effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med. 2018;379:1529–1539.
Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA-targeted therapeutics. Cell Metab. 2018;27:714–39.
Merki E, et al. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 2008;118:743–53.
Santos RD, et al. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: results of 4 phase III trials. Arterioscler Thromb Vasc Biol. 2015;35:689–99.
Merki E, et al. Antisense oligonucleotide lowers plasma levels of apolipoprotein (a) and lipoprotein (a) in transgenic mice. J Am Coll Cardiol. 2011;57:1611–21.
Nandakumar R, et al. Effects of mipomersen, an apolipoprotein B100 antisense, on lipoprotein (a) metabolism in healthy subjects. J Lipid Res. 2018;59:2397–402.
Viney NJ, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388:2239–53.
Tsimikas S, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet. 2015;386:1472–83.
Akcea Therapeutics, Inc. AKCEA-APO(a)-LRx advances as leading pharmaceutical company exercises option to license. Akcea Therapeutics, Inc. 2019. https://www.globenewswire.com/news-release/2019/02/25/1741203/0/en/AKCEA-APO-a-LRx-Advances-as-Leading-Pharmaceutical-Company-Exercises-Option-to-License.html. Accessed 4/12/2022.
HORIZON- A randomized double-blind, placebo-controlled, multicenter trial assessing the impact of lipoprotein (a) lowering with pelacarsen (TQJ230) on major cardiovascular events in patients with established cardiovascular disease (CVD). healthcare.utah.edu. https://healthcare.utah.edu/clinicaltrials/trial.php?id=FP00023345. Accessed 4/12/2022.
Tsimikas S, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382:244–55.
Lim GB. Novel siRNA reduces plasma lipoprotein(a) levels. Nat Rev Cardiol. 2022;19:147.
Korneva VA, Kuznetsova TY, Julius U. Modern approaches to lower lipoprotein(a) concentrations and consequences for cardiovascular diseases. Biomedicines. 2021;9.
Koren MJ, et al. Abstract 13951: Safety, tolerability and efficacy of single-dose Amg 890, a novel Sirna targeting Lp(a), in healthy subjects and subjects with elevated Lp(a). Circulation. 2020;142:A13951–A13951.
Kunzmann K. What’s next for lipoprotein(a) lowering agent SLN360. HCPLive https://www.hcplive.com/view/next-for-lipoprotein-a-lowering-agent-sln360. 2022.
APOLLO: Short interfering RNA shows promise for reducing lipoprotein(a). American College of Cardiology https://www.acc.org/Latest-in-Cardiology/Articles/2022/04/02/13/22/Sun-8am-APOLLO-acc-2022. Accessed 4/12/2022. The ongoing phase 1 clinical trial APOLLO is examining the effect of another siRNA, SLN360 on lowering Lp(a) levels. A single dose (300 or 600 mg) of SLN360 has been shown to produce a maximum of 96 and 98% dose reduction respectively in Lp(a) levels. The drug produces a lasting reduction of 71–81% at five months after the treatment compared to baseline Lp(a) values.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Neeja Patel, Nikita Mittal, and Parnia Abolhassan Choubdar, MD declare they have no conflict of interest. Pam R Taub, MD is a consultant for Novartis and Amgen outside the submitted work.
Human and Animal Rights Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Lipid
Rights and permissions
About this article

Cite this article
Patel, N., Mittal, N., Choubdar, P.A. et al. Lipoprotein(a)—When to Screen and How to Treat. Curr Cardiovasc Risk Rep 16, 111–120 (2022). https://doi.org/10.1007/s12170-022-00698-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12170-022-00698-8