
Abstract
The sample preparation step is very useful in obtaining precise and accurate results. Achieving a quickly performed, inexpensive, sensitive, and accurate analytical approach for measuring extremely small amounts of a dangerous compound such as mycotoxin, while posing environmental issues, has always been crucial and important. In this paper, fabric phase sorptive extraction (FPSE), a novel sorbent-based microextraction method, was used for the first time to obtain a basic and quick ochratoxin A extraction from cereals and legumes. To achieve maximum efficiency, key experimental factors in FPSE process were optimized. At the extraction stage, volume and type of solvent, time of extraction, and salt effect were evaluated. Also in the desorption step, volume and pH of back extraction solvent and time of desorption were investigated. Extracted ochratoxin A was analyzed by HPLC-FLD method. The chromatographic separation was performed using mobile phase H2O:ACN:glacial acetic acid (49:49:2, v/v/v) at a flow rate of 1.0 mL/min on C18 column with fluorescence detection (λex = 333 nm and λem = 447 nm). The detection limit was found to be 0.09 ng/g, whereas absolute acceptable recoveries (76.34–112.96%) with low relative standard deviations within day and between-day precision were (≤ 9.2% and ≤ 11.0, respectively) achieved.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mycotoxins, which are nowadays among fungal toxins, are secondary fungal metabolites and are at the center of attention (Atanda et al. 2011). Various agricultural products such as coffee and cocoa beans, soybeans, and especially cereals and legumes can contain this mycotoxin (Swanson 2011). Based on the chemical structure, ochratoxins are classified into different types of A, B, C, α, and 4-hydroxyochratoxin, among which ochratoxin A is the most toxic (Abrunhosa et al. 2010; Nogaim et al. 2020). Ochratoxin A is a relatively heat-resistant compound transmitted to the human body through the food chain (Ráduly et al. 2020). The most toxic effects of ochratoxin A are kidney toxicity (Bui-Klimke and Wu 2015), carcinogenicity (Some naturally occurring substances: food items and constituents, heterocyclic aromatic amines and mycotoxins 1993), nephrotoxic, hepatotoxic, neurotoxic, immune toxic, and also teratogenic agents (Bostan et al. 2017). Therefore, according to the World Health Organization’s motto, “healthy food is for everyone,” the responsibility of health authorities is very high.
There are many sample preparation methods used normally including solid-phase extraction (SPE) (Jahed et al. 2020; Sitterley et al. 2020; Yang et al. 2020), solid-phase microextraction (SPME) (Trujillo-Rodríguez and Anderson 2019; Zhang et al. 2018), liquid–liquid microextraction (LLME) (Arpa et al. 2018; Shishov et al. 2020), and stir bar sorptive extraction (SBSE) (Atoi et al. 2019; Khoshkbar et al. 2020; Locatelli et al. 2019b). These preparation approaches display various disadvantages, such as high consumption content of organic solvents in the conditioning as well as elution step (Kozlík et al. 2011; Zhou et al. 2013), small sorbent loading, poor sample capacity (Kabir et al. 2013a, b), considerable cost, instability, and fiber swelling in organic solvents (Nerín et al. 2009). Thus, in the current work, ochratoxin A pretreatment was prepared while adopting a new method, fabric phase sorptive extraction (FPSE), which has recently been addressed by Kabir and Furton (2013a). In this method, a fabric (polyester, fiberglass, or cellulose) is selected as the substrate, and a sol–gel sorbent is coated on its surface. An efficient extraction medium is provided by the strong covalent bond between the sol–gel and the fabric substrate. It helps the FPSE media to be exposed to any extreme chemical condition (pH, 1–13) or organic solvent without chemical/structural integrity of the microextraction device being compromised. This method has successfully addressed two main drawbacks of all sorptive extraction methods, including longer sample preparation time and low sample capacity (Kabir and Furton 2020).
FPSE has been used to analyze a broad spectrum of analytes, for instance, non-steroidal anti-inflammatory medicines (Tartaglia et al. 2020), triazine herbicides (Manousi et al. 2022a; Manousi et al. 2022b), emerging contaminants (Agadellis et al. 2020; Roldán-Pijuán et al. 2015), amphenicols (Samanidou et al. 2015), androgens/progestogens (Guedes-Alonso et al. 2016), residual sulphonamides (Karageorgou et al. 2016), benzodiazepines (Samanidou et al. 2016), alkyl phenols (Kumar et al. 2015), PAHs (Gazioglu et al. 2021a, 2021b), UV stabilizers or plastic additives (Locatelli et al. 2019a; Montesdeoca-Esponda et al. 2015), and aflatoxins (Noori et al. 2023). They could be found in various matrices, for instance, water, milk, and biological liquids (Kabir et al. 2018; Locatelli et al. 2020; Locatelli et al. 2021; Locatelli et al. 2018).
The current study aimed to explicate an FPSE-HPLC-FLD method for the ochratoxin A quick isolation in different foodstuffs, including cereals and legumes, through an optimized method for sample preparation procedure. Finally, taking into consideration the principles of green chemistry, we reached a fast, simplified, and practical approach for ochratoxin A analysis.
Materials and Methods
Chemicals
FPSE media substrates were obtained from Ardakan textile company (Yazd, Iran). Polytetrahydrofuran, methyltrimethoxysilane (MTMS), acetone, dichloromethane, sodium hydroxide, hydrochloric acid, HPLC grade acetonitrile, and methanol were purchased from Merck (Darmstadt, Germany). Trifluoroacetic acid (TFA) and glacial acetic acid were obtained from Carlo Erba (Milan, Italy) and were used without further purification. Ochratoxin A powder was prepared from Sigma (St. Louis, USA). In all the experiments, double-distilled water was used.
Instrumentation and Chromatographic Conditions
Different centrifugation solutions were performed to achieve solutions free of particles in an ortoalresa centrifuge model Digicen 21(ortoalresa, Madrid, Spain). A Fisher scientific digital vortex mixer (Fisher Scientific, USA) was used to mix different solutions thoroughly. A 2510 BRANSON ultrasonic cleaner (Branson Inc., USA) was used to provide sol solution free of bubbles. Deionized water (18.2 M) was obtained by a homemade deionized water system (Iran) for sol–gel synthesis. An AVATAR FT-IR spectrometer equipped with a universal ATR sampling accessory (Thermo, USA) was applied to carry out FT-IR characterization of fabric substrates, and sol–gel coated FPSE media. An LMU TESCAN BRNO- Mira3 field emission scanning electron microscope (Oxford Instrument, England) equipped with an EDAX detector was employed to obtain SEM images of coated and uncoated FPSE media. HPLC analyses were performed on a Waters Breeze (Waters, Milford, MA, USA), HPLC system equipped with a binary HPLC pump (Waters 1525), with attached 20-µL Rheodyne sample injector and a fluorescence detector with 333-nm excitation and 443-nm emission wavelength. The reverse-phase column was a Waters Nova-pak® C18 analytical column (250 × 4.6 mm); the temperature was adjusted to 30 °C. The mobile phase and flow rate were acetonitrile:water:glacial acetic acid (49:49:2, v/v/v) and 1 mL/min, respectively. Breeze software was used to analyze and collect the data as well as control the system operation.
Standard and Sample Preparation
To prepare ochratoxin A stock solution (1000 ng/mL), its solid standard was dissolved in methanol. The stock standard solutions were kept at − 20 °C and working standard solutions at 4 °C. Spectrophotometrical check was performed to assess the stock solution stability. Shortly before use, all working standard solutions were diluted and prepared. Samples were purchased from Mashhad (Khorasan, Iran) and tested for no contamination. A required specimen size of 1000 g was applied; then, they underwent 1-min mixing; 500 g of each sample was crushed into a fine powder with the resulting particle size of below 0.3 mm. The samples were stored at − 20 ºC for later analysis. To determine ochratoxin A, 5-g ground samples was spiked with a determined volume of ochratoxin A stock standard solution and stored for 30 min in the dark. After this time, the sample was mixed with 20 mL methanol:water (50:50, v/v) containing 5% (w/v) NaCl salt and stirred for 30 min. Once mixing was performed, a filter paper was used to filter (Whatman No. 4) the slurry, and 10 mL of this mixture was used for the next step in the fabric phase sorptive extraction method.
Preparation of Sol–Gel Polytetrahydrofuran (Poly-THF) Coated FPSE Media
Fabrics Pretreatment for Sol–Gel Coating
One critical step to prepare the sol–gel sorbent coating is the selection and pretreatment of fabric (Kabir and Furton 2020). Due to the nature and polarity of the extracted target analyte, cotton fabric (100% cellulose) was chosen for sol–gel sorbent coating as the substrate. A significant disadvantage of commercial fabric is that they contain some finishing materials to impart glossiness and keep the fiber from collecting dust particles. To optimize the sorbent loading capacity on the fabric substrate, we need to remove these residuals and make the surface hydroxyl groups active before the sol–gel sorbent coating on the fabric surface. A fabric pretreatment was necessary for cleaning it and activating hydroxyl groups. This process has been described elsewhere in detail (Kabir et al. 2017; Kumar et al. 2014).
Sol Solution Preparation for Coating on the Substrate and the Matrix Surface
The next step is to maximize the selectivity of the sol solution toward the target analyte found in the sample matrices. The nature of the sample matrix and target analyte determines how the sol solution needs to be designed. The selection of the organic polymer and sol–gel precursor has gained special attention. Selecting a proper polymer is vital for a better sorbent design; thus, the type of organic polymer is highly important in the final selection of the FPSE membrane (Kabir and Furton 2020). To be coated to 100% cotton cellulose fabric, a sol solution was prepared using poly-THF as the polymer, methyltrimethoxysilane (MTMS) as the sol–gel precursor, trifluoroacetic acid (containing 5% water) as the sol–gel catalyst, and methylene chloride:acetone (50:50, v/v) as the organic solvent.
The sol–gel coating and sol solution preparation process was described in detail elsewhere (Kumar et al. 2014). After the synthesis of the sol solution, the pretreated clean fabric was carefully put into it and kept in the reaction bottle for 4 h. Since the solvent has to be evaporated and the sol–gel coating needs aging, after removing the substrate from the solution, it was kept inside the desiccator overnight. Finally, methylene chloride:acetone (50:50, v/v) mixture was used to rinse the FPSE membrane and sonicated for 30 min. Then, it was air-dried for 1 h and kept inside an airtight packaging for later use.
Statistical Analysis
All experiments were performed three times. The statistical analysis was done using GraphPad Prism 8 and Excel 2010 software. Differences between sample groups were evaluated by GraphPad prism, with all graphs being plotted using GraphPad prism and Excel. A 0.05 level of significance was employed.
Results and Discussion
The Substrate and Sol–Gel Poly-THF Coating Chemistry
There are three building blocks for fabric phase sorptive extraction membranes: (1) a sol–gel active inorganic or organic polymer; (2) a fabric substrate; (3) a sol–gel inorganic precursor/organically modified inorganic precursor (Kabir and Furton 2020). The FPSE membrane is mainly built by fabric substrate. The general selectivity of the FPSE membrane is affected by the substrate through hydrophilic/hydrophobic interactions (Lioupi et al. 2019). Since cellulose fabric has abundant sol–gel active functional groups compared to other fabric, there is a great deal of sorbent loading per unit on cellulose fabric (Kabir and Furton 2020). Due to the medium polar nature of the selected target analyte, hydrophilic cotton cellulose seems to be a suitable substrate. Inorganic sol–gel precursor establishes sol–gel sorbent in 3D networks and exhibits a linking role to bind the networks with the fabric substrate. So it can be said that it is central to the sol–gel sorbent coating process (Celeiro et al. 2020). The selectivity and extraction affinity regarding the target analyte directly depend on sol–gel active organic polymers. Due to the structure of polymer used in the sol–gel coating reaction, various intermolecular interactions were created (Lioupi et al. 2019). As the selected analyte possesses medium polarity, we chose polytetrahydrofuran (poly-THF), a medium polar polymer containing repeating units of tetramethylene oxide and terminal hydroxyl groups the organic polymer. Moreover, trifluoroacetic acid (with 5% v/v water), methylenechloride:acetone (50/50, v/v), and methyltrimethoxysilane (MTMS) were utilized as sol–gel catalyst, solvent system, and the inorganic precursor, respectively.
Hydroxyl groups are formed as a result of the three-methoxy groups in MTMS being hydrolyzed during catalytic hydrolysis and start creating a 3D network through polycondensation. Sol–gel active organic polymer incorporates in into the network randomly when the 3D network is forming. Eventually, as a result of polycondensation, there is a chemical bond between the sol–gel sorbent network and the fabric substrate via hydroxyl groups of the fabric (Kabir and Furton 2020). As the sol–gel sorbent is very porous and the fabric substrate is so permeable, the aqueous sample can travel through the FPSE membrane countlessly during the process of extraction and guarantees rather full extraction of the target compound within a reasonably short time (Alampanos et al. 2019).
Characterization of Sol–Gel PTHF Coated FPSE Media
Scanning Electron Microscopy
The characterization of sol–gel sorbent-coated fabric phase sorptive extraction membranes is often done in different ways, one of which is scanning electron microscopy (SEM). SEM image reflects important morphological properties of the fabric substrate surface. SEM was used to compare the surface morphology before and after the coating process; Fig. 1 shows SEM micrographs of (a) the surface of uncoated cellulose fabric substrate and (b) sol–gel poly-THF coated fabric phase sorptive extraction media. An SEM image reveals numerous micro-fibrils as well as bundles of micro-fibrils integrated to possess well-structured macropores even after sol–gel PTHF coating. These macropores cause faster and near exhaustive extraction kinetic which allow the sample matrix easily flow as the analyte extraction is performing. The SEM images show homogeneous and sponge-like porous of thin PTHF sorbent coating around the cellulose micro-fibrils which in turn provides quicker permeation of the sample matrix. It contains the target analyte and eventually leads to shortening the extraction time of equilibrium (Celeiro et al. 2020).

SEM images of (a) uncoated sample of FPSE medium with surface micro-fibrils and pores surface; (b) uniformly coated sol–gel poly-THF onto the fabric matrix
FT-IR Spectroscopy
FT-IR spectra provide information on how various sol solution components chemically integrate into the sol–gel sorbent network and the fabric substrate. Figure 2 shows two FT-IR spectra: uncoated cellulose substrate (a) and sol–gel poly-THF coated FPSE media (b). Characteristic absorption in two FT-IR spectra appears in three peak areas between 3000 and 3400 cm−1; between 1307 and 2900 cm1; and at 1090 cm−1 which correspond to O–H, C–H, and C–O bending vibration. The indication of a transparent band in the FT-IR spectrum at 1096 cm−1 is that Si–O–C bonds could have established from the successfully incorporated sol–gel poly-THF network into the cellulose substrate. Also, regarding the systematic reduction in the absorption bands between 3000 and 3400 cm−1 in FT-IR spectrum of sol–gel coated FPSE media correspond to O–H bending vibrations. It can be said that this reduction is due to the involvement of hydroxyl groups of cellulose substrate to the sol–gel network (Montarsolo et al. 2013). There is no other clear signal of sol–gel poly-THF coating as absorption bands overlaps with those of uncoated cellulose fabric. The sharpest peak in FT-IR spectra of sol–gel coated FPSE membrane compared with cellulose substrate shows the uniform coverage of sol–gel around the cellulose substrate that can be seen in the SEM image.

FT-IR spectrum of uncoated cellulose substrate (a); sol–gel poly-THF coated FPSE medium (b)
FPSE Procedure
The fabric phase sorptive extraction protocol involves three steps: (a) cleaning of FPSE membrane, (b) extraction, (c) back extraction (Kabir and Furton 2020). Before the extraction, the FPSE membrane was conditioned for its use. This work is done by immersing the FPSE membrane into 2 mL of methanol:acetonitrile (50:50, v/v) for 5 min, and subsequently in 2 mL of ultrapure water for 3 min to remove any undesirable impurities and the remains of organic solvents. For extraction or the sorption of the target analyte by the sorbent, a clean FPSE membrane was drowned in 5 mL of the representative aliquot of the sample containing 5 ng/mL ochratoxin A. Then, the vial was sealed, and the mixture solution was magnetic stirred for 1 h so that the analyte could thoroughly be absorbed into it. Once absorption was completed, the FPSE membrane was taken from the vial and air-dried on a watch glass at room temperature. Subsequently, the back extraction solvent containing 5 mL of acetonitrile:water (50:50 v/v) solution was used for 1 h in contact with the substrate. The collected analyte was eventually centrifuged in a microtube at 13,000 rpm for 5 min to gain a solution free from particulate matters. Centriguged solution was filtered with a syringe filter (0.45 µm) and then injected into the HPLC system.
After optimizing the experimental parameters, the final FPSE conditions were set using a 5-cm2 (2.0 × 2.5 cm) FPSE device with PTHF based sol–gel coating for the extraction of 10 mL of methanol:water (50:50, v/v) containing 5% NaCl salt, for 40 min. Analyte desorption was reached using 1 mL of back extraction solution at pH = 6 for 15 min under sonication.
Optimization of Efficiency FPSE Conditions
Some key variables were studied to maximize extraction performance of analyte, including (a) extraction solvent solution, (b) the effect of addition of salt into the extraction solvent solution, (c) volume of desorption/back extraction solvent, (d) pH of desorption solvent, (e) time of desorption, (f) time of extraction, and (g) volume of extraction solvent.
Extraction Solvent Solution Effect
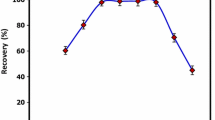
Different solvent mixtures were tried to optimize the analyte’s sorption from the sample matrix on the FPSE media. According to the obtained results (Fig. 3a), the methanol:water (50:50, v/v) mixture exhibits better performance than other mixtures. This is because the selected target analyte is medium polar. Therefore, due to the polarity, it demonstrated higher solubility in the same ratio of methanol:water in comparison with other solutions. In addition, probably this solution with respect to the FPSE membrane’s polarity can provide better conditions to help the target analyte stay in the proper state of polarity and ionic state (pka) for effective adsorption on the FPSE media. The other reason for higher efficiency of this solution—as the sorption solution—is that methanol:water (50:50, v/v) mixture has better performance to raise the activity and ability of the sol–gel network of FPSE membrane for sorption the target analyte from the sample matrix in comparison with other solution. In this step, the volume of solvent in all samples was 5 mL, and the volume of the sorption solvent was optimized in further experimentation.

Extraction solvent effect (a), effect of ionic strength (b), pH effect on desorption solvent (c), and extraction time effect (d) on ochratoxin extraction. (Error bars are standard deviation of mean)
Ionic Strength Effect
For the evaluation of sodium chloride salt effect on the adsorption attributes of ochratoxin A, some sodium chloride concentrations, including 0%, 5%, 10%, 15%, 20%, and 30% (w/v), were added to the extraction solvent solution; then, their impact on the analyte adsorption on the FPSE membrane was studied. An increase in sodium chloride salt concentration from 0 to 5% caused higher extraction efficiency. Nevertheless, as NaCl concentration reached 30%, the extraction efficiency experienced a decline (Fig. 3b). Therefore, the addition of 5% of NaCl was considered a suitable alternative. The observed difference in extraction efficiencies is probably due to the contradictory effects due to the salt added to the solution. On the other hand, the analyte gradually loses its solubility in the aqueous solution as the ionic strength improves, and a salting-out effect is produced, which makes the target analyte more available in the sorption media. It is noteworthy that viscosity increases when NaCl concentrations are high leading to the incomplete mass transfer of the analyte. This negatively affects the compound’s sorption by the FPSE media (Gülle et al. 2019).
Desorption Solvent Volume Effect
To determine the optimized volume of desorption solvent, five different volumes of desorption solvent solution (acetonitrile:water (50:50 v/v)) including 1, 2, 3, 4, and 5 mL were used and, the best result was obtained when 1 mL of desorption solvent was applied. Thus, this volume was regarded as the optimized volume in future experimentation.
Effect of pH on Desorption Solvent
The impact of solvent pH on the efficiency of desorption was investigated at pH 2, 4, 6, and 8. According to the obtained results (Fig. 3c), the optimum results were achieved under the approximately neutral condition at pH = 6. To explain this matter, it can be said that since the elution of analyte probably is due to its polarity (log Kow) and ionic state (pKa) at the elution conditions, probably in this pH value, suitable conditions were provided for the elution concerning the nature of the target analyte (Zilfidou et al. 2019). In addition, the sol–gel network of the FPSE membrane remains so active for the highest interaction with the back extraction solution. Furthermore, to keep this method as simple and easily applicable as possible in laboratory routines a moderate pH was used.
Desorption Time Effect
After the sorption step, the FPSE media was placed in contact with the desorption solvent for 5, 10, 15, 20, 25, and 30 min under sonication to obtain the optimized desorption time. A rapid increase was observed in the peak areas from 5 to 15 min. There was no significant increase in desorption value when the extraction time extended to 30 min. The equilibrium state reached for the FPSE coating media and the working solution after 15 min. Thus, the optimization time was reported as 15 min for back extraction in subsequent works.
Extraction Time Effect
The analyte’s distribution coefficients determine extraction and elution times, which create the adsorption equilibrium between the sample solution and the FPSE medium and affects the FPSE procedure performance (Zilfidou et al. 2019). To reach the maximum extraction of the analyte with FPSE membrane and evaluate the optimized extraction time, the analyte was placed in contact with the FPSE media from 10, 20, 30, 40, 50, and 60 min. The findings indicated that the extraction efficiency enhanced with time, and in 40 min, it reached the maximum value; however, no significant improvement was observed as contact time extended (Fig. 3d). Moreover, the shorter the extraction time, the more efficient the routine analysis approach. Thus, the shortest extraction time is intended. Overall, to achieve better results, 40 min extraction time was considered the optimum time for extraction.
Effect of Extraction Solvent Volume
For the optimization of the extraction solvent volume, the FPSE media was in contact with 2, 4, 6, 8, and 10 mL of a standard solution containing 5 ng/mL of ochratoxin A. The efficiency of extraction process improved as we increased the sample volume from 2 to 10 mL; therefore, 10 mL was selected as the optimized volume of the extraction solution. A high volume of extraction solution can help analyte diffusion and mass transfer through the sample matrix to the extraction media. It shortens the equilibrium creation time for making the extraction more efficient. It can be concluded that the solution in high volume can lead to greater extraction values (Alampanos et al. 2019).
Final Optimized Fabric Phase Sorptive Extraction Protocol
The FPSE media was placed in a vial with 10 mL of MeOH:H2O (50:50; v/v) at the pH = 6 containing 5% (w/v) NaCl salt and stirred for 40 min with a magnetic stir bar. For desorption step, the FPSE media was placed in vial and 1 mL of desorption solvent was added. The vial was placed in an ultrasound bath for 10 min. Afterwards, the FPSE medium was removed, and extraction solution was filtered through a syringe filter. The final extract was analyzed by HPLC-FLD.
Method Validation
A calibration curve was prepared using the optimized FPSE-HPLC conditions. The linear range of the calibration curve was 1.5–25 ng/mL, and correlation coefficients were greater than that of 0.98. The characteristics of calibration curves are presented in Table 1. The limit of detection (LOD) and the limit of quantification (LOQ) were defined as signal to noise 3:1 and 10:1, respectively. FDA requires that the intensities at LOQ must have precision of 20% at most and trueness of ± 20%.
The recovery of the ochratoxin A was used to determine the trueness. Relative recovery is distinguished as the ratio of the concentrations found in the investigated matrix to those in distilled or deionized water spiked with the exact amounts of analytes. The recovery of ochratoxin from samples was in an acceptable range (Table 2). The relative standard deviation (RSD) was within the laboratory repeatability, 3.3% (Table 1). Ochratoxin A was quantified in real samples, indicating the effectiveness of the presented method.
Application for Real Samples
Real samples, including rice and lentil, were evaluated following the spiking with ochratoxin A for validation of the developed FPSE-HPLC method. At the retention time corresponding to ochratoxin A, no interfering peak was observed during its determination in cereals or legumes samples. Acceptable recovery was obtained for rice (80.3–102.5%) and lentil (80.3–109.2%), which were in the range recommended by AOAC (Locatelli et al. 2019b). The percentage recovery from two type samples was different at various concentrations, probably due to unpredictable matrix behavior. FPSE-HPLC-FLD typical chromatograms of spiked rice samples (at the level of 5 and 10 ng/g) are depicted in Fig. 4 (R1 and R2 for 5 and 10 ng/g, respectively). The spiked lentil samples (at the level of 5 and 10 ng/g) are shown in Fig. 4 (L1 and L2 for 5 and 10 ng/g, respectively). The blank chromatograms for rice and lentil are shown in Fig. 4 (RB and LB). The determination results are given in Table 2.

Typical FPSE-HPLC-FLD chromatograms of spiked rice samples at the level of 5 ng/g (R1) and 10 ng/g (R2) and lentil samples at the level of 5 ng/g (L1) and 10 ng/g (L2). The RB and LB are blank chromatogram for rice and lentil samples, respectively
Estimation of Uncertainty
Uncertainty is defined by ISO as a parameter. It is a range of possible values in which true measures have been lied. These values could reasonably be attributed to the relevant measurement. Parameters that affect uncertainty are volume, mass, calibration curve, and purity of the reference material (ISO 170252017). In general, the uncertainty of measurements was consist of many components, for example, in an analytical result sampling technique, extraction, clean-up, and instrumental determination could be interfered. Table 2 shows the value of uncertainty for ochratoxin A in rice and lentil samples used in the present study at the spiked levels.
Comparison with Other Reported Methods
Characterization of ochratoxin A in food samples by FPSE-HPLC-FLD was compared to some microextraction techniques applied in food and biological samples, such as DLLME-LC–MS/MS, SPME-HPLC-FLD, HF-LPME-HPLC-FLD, SPME-LC–MS/MS, and MI-μSPE-HPLC-FLD (Table 3) (Campone et al. 2011; González-Peñas et al. 2004; Iqbal et al. 2016; Lee et al. 2012; Vatinno et al. 2008a, 2008b). Since the cotton fabric coated PTHF displays higher mass transfer of analyte to the coating surface from the bulk phase, it can be concluded that our method needs considerably shorter extraction time in comparison with various current methods in this area. Data regarding the RSD and recovery obtained here can simply be compared with those from previous methods. Moreover, the calculated LOD was acceptable compared to other approaches. These findings reveal that our novel FPSE-HPLC-FLD method is a quick, convenient, and highly efficient way that can be readily used to determine ochratoxin A in food samples, without harsh extraction conditions and time-consuming protein precipitation steps..
Conclusion
Measurement of compounds, elements, and toxins in trace amounts—in addition to environmental issues—has always been necessary and important in aquatic, biological, food samples, etc. Sample preparation is usually regarded as a sophisticated and demanding stage, especially when the analyte concentration level is trace/ultra-trace in environmental matrices. There has been a growing trend toward sensitivity and miniaturization of analytical methods. One way to achieve this goal is to use rapid, inexpensive, sensitive, and accurate analytical methods to measure target trace amounts. The sample preparation step in the analytical methods often involves an extraction step to separate and concentrate the target analyte from the sample matrix. Choosing the method and extraction conditions has a special role in the analysis time, easiness of application, and selectivity. In this research, we developed and validated a novel FPSE-HPLC-FLD approach to extract/pre-concentrate ochratoxin A in food samples. The results show that fabric phase sorptive extraction method can be effective in reducing solvent volume and analysis time, making this method more economic, green, and environmentally friendly. This method had high validate and experienced the range, linearity, and at the same time was precise and accurate. Therefore, this study demonstrates that the FPSE method displays acceptable levels of speed, precision, reproducibility, and sensitivity for determining ochratoxin A in food samples.
Data Availability
The datasets applied and analyzed during this research are available from the corresponding author on request.
Abbreviations
- ACN :
-
Acetonitrile
- DLLME :
-
Dispersive liquid–liquid microextraction
- FPSE :
-
Fabric phase sorptive extraction
- HF :
-
Hollow fiber
- HOAC :
-
Acetic acid
- HPLC-FLD :
-
High-performance liquid chromatography-fluorescence detection
- LOD :
-
Limit of detection
- LOQ :
-
Limit of quantification
- LPME :
-
Liquid-phase micro extraction
- MeOH :
-
Methanol
- MTMS :
-
Methyltrimethoxysilane
- PTHF :
-
Polytetrahydrofuran
- RSD :
-
Relative standard deviation
- SEM :
-
Scanning electron microscopy
- SPE :
-
Solid-phase extraction
- SPME :
-
Solid-phase microextraction
- WHO :
-
World Health Organization
References
Abrunhosa L, Paterson RR, Venâncio A (2010) Biodegradation of Ochratoxin A for Food and Feed Decontamination Toxins 2:1078–1099
Agadellis E, Tartaglia A, Locatelli M, Kabir A, Furton KG, Samanidou V (2020) Mixed-mode fabric phase sorptive extraction of multiple tetracycline residues from milk samples prior to high performance liquid chromatography-ultraviolet analysis. Microchemical Journal 159:105437. https://doi.org/10.1016/j.microc.2020.105437
Alampanos V, Kabir A, Furton KG, Samanidou V, Papadoyannis I (2019) Fabric phase sorptive extraction for simultaneous observation of four penicillin antibiotics from human blood serum prior to high performance liquid chromatography and photo-diode array detection. Microchem J 149:103964
Arpa Ç, Albayati S, Yahya M (2018) Effervescence-assisted dispersive liquid-liquid microextraction based on deep eutectic solvent for preconcentration and FAAS determination of copper in aqueous samples. Int J Environ Anal Chem 98:938–953
Atanda S, Pessu P, Agoda S, Isong I, Adekalu O, Echendu M, Falade T (2011) Fungi and mycotoxins in stored foods. African Journal of Microbiology Research 5:4373–4382
Atoi PA, Talebpour Z, Fotouhi L (2019) Introduction of electropolymerization of pyrrole as a coating method for stir bar sorptive extraction of estradiol followed by gas chromatography. J Chromatogr A 1604:460478
Bostan HB, Danesh NM, Karimi G et al (2017) Ultrasensitive detection of ochratoxin A using aptasensors. Biosens Bioelectron 98:168–179
Bui-Klimke TR, Wu F (2015) Ochratoxin A and human health risk: a review of the evidence. Crit Rev Food Sci Nutr 55:1860–1869
Campone L, Piccinelli AL, Rastrelli L (2011) Dispersive liquid-liquid microextraction combined with high-performance liquid chromatography-tandem mass spectrometry for the identification and the accurate quantification by isotope dilution assay of ochratoxin A in wine samples. Analytical and Bioanalytical Chemistry 399(1279):1286. https://doi.org/10.1007/s00216-010-4347-7
Celeiro M, Acerbi R, Kabir A, Furton KG, Llompart M (2020) Development of an Analytical Methodology Based on Fabric Phase Sorptive Extraction Followed by Gas Chromatography-Tandem Mass Spectrometry to Determine UV Filters in Environmental and Recreational. Waters Analytica Chimica Acta X 4:100038
Gazioglu I, Kabir A, Zengin OS, KepekciTekkeli E, Furton KG, Tartaglia A, Locatelli M (2021) Development of sol-gel phenyl/methyl/poly (dimethylsiloxane) sorbent coating for fabric phase sorptive extraction and its application in monitoring human exposure to selected polycyclic aromatic hydrocarbons using high performance liquid chromatography-fluorescence detection. Journal of Chromatography B 1163:122520. https://doi.org/10.1016/j.jchromb.2020.122520
Gazioglu I, Zengin OS, Tartaglia A, Locatelli M, Furton KG, Kabir A (2021b) Determination of polycyclic aromatic hydrocarbons in nutritional supplements by fabric phase sorptive extraction (FPSE) with high performance liquid chromatography (HPLC) with fluorescence detection. Anal Lett 54:1683–1696 https://doi.org/10.1080/00032719.2020.1821209
González-Peñas E, Leache C, Viscarret M (2004) Pérez de Obanos A, Araguás C, López de Cerain A. Determination of Ochratoxin A in Wine Using Liquid-Phase Microextraction Combined with Liquid Chromatography with Fluorescence Detection Journal of Chromatography A 1025:163–168. https://doi.org/10.1016/j.chroma.2003.10.113
Guedes-Alonso R, Ciofi L, Sosa-Ferrera Z, Santana-Rodríguez JJ, Del Bubba M, Kabir A, Furton KG (2016) Determination of androgens and progestogens in environmental and biological samples using fabric phase sorptive extraction coupled to ultra-high performance liquid chromatography tandem mass spectrometry. J Chromatogr A 1437:116–126
Gülle S, Ulusoy HI, Kabir A, Tartaglia A, Furton KG, Locatelli M, Samanidou VF (2019) Application of a fabric phase sorptive extraction-high performance liquid chromatography-photodiode array detection method for the trace determination of methyl paraben propyl paraben and butyl paraben in cosmetic and environmental samples. Analytical Methods 11:6136–6145. https://doi.org/10.1039/C9AY02260K
Iqbal SZ, Asi MR, Hanif U, Zuber M, Jinap S (2016) The presence of aflatoxins and ochratoxin A in rice and rice products and evaluation of dietary intake. Food Chemistry 210:135–140. https://doi.org/10.1016/j.foodchem.2016.04.104
ISO 17025 2017 General requirements for the competence of testing and calibration laboratories
Jahed FS, Hamidi S, Ghaffary S, Nejati B 2020 Dispersive micro solid phase extraction of busulfan from plasma samples using novel mesoporous sorbent prior to determination by HPLC-MS/MS Journal of Chromatography B 122091
Kabir A, Furton KG, Malik A (2013b) Innovations in sol-gel microextraction phases for solvent-free sample preparation in analytical chemistry. TrAC, Trends Anal Chem 45:197–218
Kabir A, Mesa R, Jurmain J, Furton KG (2017) Fabric Phase Sorptive Extraction Explained Separations 4:21
Kabir A, Furton KG, Tinari N et al. (2018) Fabric phase sorptive extraction-high performance liquid chromatography-photo diode array detection method for simultaneous monitoring of three inflammatory bowel disease treatment drugs in whole blood plasma and urine. J Chromatogr B Analyt Technol Biomed Life Sci 1084:53–63. https://doi.org/10.1016/j.jchromb.2018.03.028
Kabir A, Furton K 2013a Fabric phase sorptive extractors (FPSE) US Patent Application 14 US20140274660A20140274661
Kabir A, Furton KG 2020 Fabric phase sorptive extraction a new genration green sample preparation approach In Solid-phase extraction Handbooks in separation science Elsevier pp 355–386
Karageorgou E, Manousi N, Samanidou V, Kabir A, Furton KG (2016) Fabric phase sorptive extraction for the fast isolation of sulfonamides residues from raw milk followed by high performance liquid chromatography with ultraviolet detection. Food Chem 196:428–436
Khoshkbar ZN, Talebpour Z, Ghamat SN, Farzaneh F 2020 Fabrication, characterization and application of zeolitic imidazolate framework-8/acrylate monolithic polymer as a composite phase for stir bar sorptive extraction Microchemical Journal 105060
Kozlík P, Bosáková Z, Tesařová E, Coufal P, Čabala R (2011) Development of a solid-phase extraction with capillary liquid chromatography tandem mass spectrometry for analysis of estrogens in environmental water samples. J Chromatogr A 1218:2127–2132
Kumar R, Kabir A, Furton KG, Malik AK (2015) Development of a fabric phase sorptive extraction with high-performance liquid chromatography and ultraviolet detection method for the analysis of alkyl phenols in environmental samples. J Sep Sci 38:3228–3238
Kumar R, Malik AK, Kabir A, Furton KG 2014 Efficient analysis of selected estrogens using fabric phase sorptive extraction and high performance liquid chromatography-fluorescence detection Journal of Chromatography A 1359 16–25
Lee TP, Saad B, Ng EP, Salleh B (2012) Zeolite Linde Type L as micro-solid phase extraction sorbent for the high performance liquid chromatography determination of ochratoxin A in coffee and cereal. J Chromatogr A 1237:46–54. https://doi.org/10.1016/j.chroma.2012.03.031
Lioupi A, Kabir A, Furton KG, Samanidou V (2019) Fabric phase sorptive extraction for the isolation of five common antidepressants from human urine prior to HPLC-DAD analysis. J Chromatogr B 1118:171–179
Locatelli M, Tinari N, Grassadonia A et al. (2018) FPSE-HPLC-DAD method for the quantification of anticancer drugs in human whole blood plasma and urine. Journal of Chromatography B 1095:204–213. https://doi.org/10.1016/j.jchromb.2018.07.042
Locatelli M, Furton KG, Tartaglia A, Sperandio E, Ulusoy HI, Kabir A (2019) An FPSE-HPLC-PDA method for rapid determination of solar UV filters in human whole blood plasma and urine. J Chromatogr B Analyt Technol Biomed Life Sci 1118–1119:40–50. https://doi.org/10.1016/j.jchromb.2019.04.028
Locatelli M et al (2019b) Innovative configurations of sample preparation techniques applied in bioanalytical chemistry: a review. Curr Anal Chem 15:731–744. https://doi.org/10.2174/1573411015666190301145042
Locatelli M, Tartaglia A, D’Ambrosio F, Ramundo P, Ulusoy HI, Furton KG, Kabir A (2020) Biofluid sampler a new gateway for mail-in-analysis of whole blood samples. Journal of Chromatography B 1143:122055. https://doi.org/10.1016/j.jchromb.2020.122055
Locatelli M et al (2021) Fabric-phase sorptive membrane array as a noninvasive in vivo sampling device for human exposure to different compounds. Anal Chem 93:1957–1961. https://doi.org/10.1021/acs.analchem.0c04663
Manousi N et al (2022) Exploring sol-gel zwitterionic fabric phase sorptive extraction sorbent as a new multi-mode platform for the extraction and preconcentration of triazine herbicides from juice samples. Food Chemistry 373:131517. https://doi.org/10.1016/j.foodchem.2021.131517
Manousi N, Kabir A, Furton KG, Zachariadis GA, Rosenberg E (2022) Expanding the applicability of magnet integrated fabric phase sorptive extraction in food analysis extraction of triazine herbicides from herbal infusion samples. Microchemical Journal 179:107524. https://doi.org/10.1016/j.microc.2022.107524
Montarsolo A, Periolatto M, Zerbola M, Mossotti R, Ferrero F (2013) Hydrophobic sol-gel finishing for textiles: improvement by plasma pre-treatment. Text Res J 83:1190–1200
Montesdeoca-Esponda S, Sosa-Ferrera Z, Kabir A, Furton KG, Santana-Rodríguez JJ (2015) Fabric phase sorptive extraction followed by UHPLC-MS/MS for the analysis of benzotriazole UV stabilizers in sewage samples. Anal Bioanal Chem 407:8137–8150
Nerín C, Salafranca J, Aznar M, Batlle R (2009) Critical review on recent developments in solventless techniques for extraction of analytes. Anal Bioanal Chem 393:809
Nogaim QA, Bugata LSP, Prabhakar P, Reddy UA, Kumari I, Mahboob M (2020) Protective Effect of Yemeni Green Coffee Powder against the Oxidative Stress Induced by Ochratoxin A Toxicology Reports 7:142–148
Noori H, Feizy J, Eshaghi Z (2023) Development of a Nanoparticle-Assisted Fabric Phase Sorptive Extraction Technique Coupled with High-Performance Liquid Chromatography for Sensitive Determination of Aflatoxins in Food Samples. Analytical and Bioanalytical Chemistry Research 10:121–134. https://doi.org/10.22036/abcr.2022.353555.1801
Ráduly Z, Szabó L, Madar A, Pócsi I, Csernoch L (2020) Toxicological and medical aspects of Aspergillus-derived mycotoxins entering the feed and food chain. Front Microbiol 10:2908
Roldán-Pijuán M, Lucena R, Cárdenas S, Valcárcel M, Kabir A, Furton KG (2015) Stir fabric phase sorptive extraction for the determination of triazine herbicides in environmental waters by liquid chromatography. J Chromatogr A 1376:35–45
Samanidou V, Galanopoulos L-D, Kabir A, Furton KG (2015) Fast extraction of amphenicols residues from raw milk using novel fabric phase sorptive extraction followed by high-performance liquid chromatography-diode array detection. Anal Chim Acta 855:41–50
Samanidou V, Kaltzi I, Kabir A, Furton KG (2016) Simplifying sample preparation using fabric phase sorptive extraction technique for the determination of benzodiazepines in blood serum by high-performance liquid chromatography. Biomed Chromatogr 30:829–836
Shishov A, Gerasimov A, Nechaeva D, Volodina N, Bessonova E, Bulatov A 2020 An effervescence-assisted dispersive liquid-liquid microextraction based on deep eutectic solvent decomposition determination of ketoprofen and diclofenac in liver Microchemical Journal 104837
Sitterley KA, Linden KG, Ferrer I, Thurman EM 2020 Desalting and concentration of common hydraulic fracturing fluid additives and their metabolites with solid-phase extraction Journal of Chromatography A 461094
Some naturally occurring substances: food items and constituents, heterocyclic aromatic amines and mycotoxins 1993 WHO IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans 56
Swanson KM 2011 Nuts oilseeds dried legumes and coffee In Microorganisms in foods Springer 227–239
Tartaglia A et al (2020) Fast off line FPSE HPLC PDA determination of six NSAIDs in saliva samples. Journal of Chromatography B 1144:122082. https://doi.org/10.1016/j.jchromb.2020.122082
Trujillo-Rodríguez MJ, Anderson JL (2019) Silver-based polymeric ionic liquid sorbent coatings for solid-phase microextraction: materials for the selective extraction of unsaturated compounds. Anal Chim Acta 1047:52–61
Vatinno R, Aresta A, Zambonin CG, Palmisano F (2008a) Determination of ochratoxin A in green coffee beans by solid-phase microextraction and liquid chromatography with fluorescence detection. J Chromatogr A 1187:145–150
Vatinno R, Vuckovic D, Zambonin CG, Pawliszyn J (2008) Automated high-throughput method using solid-phase microextraction-liquid chromatography-tandem mass spectrometry for the determination of ochratoxin A in human urine. Journal of Chromatography A 1201:215–221. https://doi.org/10.1016/j.chroma.2008.05.079
Yang H, Li L, Cao H, Zhang Z, Zhao T, Hao Y, Wang M 2020 Silica supported metal organic framework 808 composites as adsorbent for solid-phase extraction of benzodiazepines in urine sample Microchemical Journal 105062
Zhang L, Gionfriddo E, Acquaro V Jr, Pawliszyn J (2018) Direct immersion solid-phase microextraction analysis of multi-class contaminants in edible seaweeds by gas chromatography-mass spectrometry. Anal Chim Acta 1031:83–97
Zhou F et al (2013) Measurement of phenolic environmental estrogens in human urine samples by HPLC-MS/MS and primary discussion the possible linkage with uterine leiomyoma. Journal of Chromatography B 938:80–85
Zilfidou E, Kabir A, Furton KG, Samanidou V (2019) An improved fabric phase sorptive extraction method for the determination of five selected antidepressant drug residues in human blood serum prior to high performance liquid chromatography with diode array detection. J Chromatogr B 1125:121720
Acknowledgements
The authors would like to express their gratitude to the Research Institute of Food Science and Technology (RIFST) and Payam Noor University.
Author information
Authors and Affiliations
Contributions
AE: did the laboratory work, wrote the draft of this report, reviewed the final version of this report, and supplied all figures and tables. AM: supervised, designed, and directed the project. JF: supervised, conceived, and planned the experiments wrote the final version of this report; and evaluated all data. All the authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors stated that there are no conflicts of interest regarding the publication of this article
Ethics Approval and Consent to Participate
Human samples have not been used in this study.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Olia, A.E.A., Mohadesi, A. & Feizy, J. A Fabric Phase Sorptive Extraction Protocol Combined with Liquid Chromatography-Fluorescence Detection for the Determination of Ochratoxin in Food Samples. Food Anal. Methods 16, 974–984 (2023). https://doi.org/10.1007/s12161-023-02474-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-023-02474-9