
Abstract
Multihalo anisoles (MHAs) bring significant loss in wine industry due to their unpleasant taste and potential health concern. They are the methylation derivatives of multihalo phenols (MHPs) and transferred from corks or barrels in wine production and storage process. Most of previous reports targeted on 2,4,6-trichloroanisole (TCA), the most common one of MHAs. Here, we report a method which could detect nine kinds of MHAs and MHPs simultaneously. Acetic anhydride was chosen as derivative reagent to transfer phenols to their acetates which are essential for the following solid phase microextraction and chromatographic separation. Head space solid phase microextraction (HS-SPME) condition were optimized as 5 mL of wine sample, 1 g of sodium chloride, 5 min of equilibrium time, 60 ºC of extraction temperature, and 45 min of extraction time. Gas chromatography tandem mass spectrometry was selected for separation, detection, and quantification of the target analytes. The limit of quantifications (LOQs), limit of detections (LODs), linear range, recovery yield, and relative standard deviation of nine kinds of MHAs and MHPs were also investigated. This method was validated by measuring MHPs and MHAs simultaneously in commercial wine samples produced in different countries.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Wine is the natural alcoholic drink produced by the fermentation of grape juice and one of the most popular beverages around the world (Waterhouse et al. 2016; Finley 2018). There are lots of active components in wine with the functions of anti-oxidation, disease preventing, intestinal colony regulating etc. (Bertelli 2007; Urquiaga et al. 2010; Chen et al. 2010; Requena et al. 2010). There is consensus that moderate wine consumption is good for health.
However, off flavor or cork taint in wine is one of the major problems spoil the quality and acceptability of wine, which would cause significant economic loss and reputation damage of winery (Riu et al. 2002; Sefton et al. 2005; Fontana, 2012; Vlachos et al. 2007). The most concerned off flavor in wine is the MHAs either from corks or from barrels during wine production, cardboard packing, and storage. Phenols, such as TCP, PeCP or TBP, are the well-known environmental pollutants, which are used as pesticides, disinfectants, or retardants in the plant growth or cork producing process (Alvarez-Rodriguez et al. 2003; Simpson et al. 2007; Waterhouse et al. 2016). MHPs are the substituted products of phenols and halogen sources via electrophilic substitution reaction (Waterhouse et al. 2016), they also could be the degradation products of lindan via microbiological dehalogenation (Pollnitz et al. 1996). They could be methylated to form anisoles by fungi (Coque et al. 2003; Insa et al. 2004). The taste of these pollutants has strong unpleasant, mold, or rotten-wood smell at trace concentration. It is reported that the organoleptic and rejection threshold of 2,4,6-trichloro anisole (TCA) in Chardonnay wine are as low as 2.1 ng/L and 3.1 ng/L respectively (Hjelmeland et al. 2012; Vlachos et al. 2007). Although most previous reports about off flavors focus on the TCA, other MHAs are also studied. For examples, 2,4,6-tribromo anisole (TBA) has the threshold concentration similar to TCA, while 2,3,4,6-tetrachloro anisole (2,3,4,6-TeCA) and pentachloro anisole (PeCA) have the higher threshold concentrations (Boutou et al. 2007). MHPs, regarded as one of the precursors of MHAs, could also cause off flavors and health issues. For instances, pentachloro phenol (PeCP) is on the list of International Agency for Research on Cancer as Group I (carcinogenic to human); TCP belongs to Group 2B (possibly carcinogenic to human) (IARC working group 2016); PeCP and 2,4,6-tricholro phenol (TCP), widely used as wood preservative for decades, have been regarded as priority environmental pollutants (Zhu et al. 2017). Therefore, MHPs in wine should also be paid attention to for safety and quality concerns.
Due to the trace concentration of MHAs and/or MHPs in wine or cork stoppers, the sample preparation techniques are crucial and have been investigated in lots of publications. For instances, Lizarraga E et al. applied HS-SPME for the determination of TCA, TeCA, and PeCA in red wine (Lizarraga et al. 2004). Salinas and co-workers extract MHAs and MHPs in wine via stir bar sorptive extraction (SBSE) (Zalacain et al. 2004). Hernández-Córdoba group conducted dispersive liquid–liquid microextraction (DLLME) to extract chlorophenols and haloanisoles in wines and corks (Campillo et al. 2010). Pizarro and co-works used solid phase extraction (SPE) and silylation-HS-SPME to determine MHAs and MHPs in red wine (Martinez-Urunuela et al. 2005; Pizarro et al. 2007). A supercritical fluid extraction (SFE) method was developed by Ebeler group to extract TCA from cork stoppers (Taylor et al. 2000). Tena et al. applied accelerated solvent extraction (ASE) or pressurized fluid extraction (PFE) to extract TCA from cork stoppers (Ezquerro et al. 2006). The successful identification and determination of MHAs and/or MHPs in wine or cork stoppers through different extraction techniques clearly demonstrates the importance of sample preparation techniques.
Gas chromatography, a powerful separation technique with good separation efficiency, is one of the most widely used chromatographic methods in quantification of small organic analytes. It is very versatile and can be tandemed with different high sensitive detectors to determine trace amount targets in matrix, such as electron captured detector (ECD) (Insa et al. 2004; Vlachos et al. 2007), MS (Zalacain et al. 2004; Lizarraga et al. 2004; Campillo et al. 2010; Ezquerro et al. 2006), and tandem MS (MS/MS) (Martinez-Urunuela et al. 2005; Hjelmeland et al. 2012).
Based on the above mentioned sample preparation, separation, and detection techniques, there are numerous combination of the method development to determine MHAs and/or MHPs in wines or cork stoppers. Here, we report a quantitative method to determine two classification pollutants (MHAs and MHPs) in wine simultaneously by online derivatization HS–SPME–GC–MS/MS.
Materials and Methods
Chemicals and Samples
Standard grade chemicals of 2,4,6-trichloro anisole (TCA), 2,3,4,6-tetrachloro anisole (2,3,4,6-TeCA), 2,3,4,5-tetrachloro anisole (2,3,4,5-TeCA), 2,4,6-tribromo anisole (TBA), 2,4,6-trichloro phenol (TCP), 2,3,4,6-tetrachloro phenol (2,3,4,6-TeCP), and pentachloro anisole (PeCA), 2,4,6-tribromo phenol (TBP) were purchased from Dr. Ehrenstorfer. Pentachloro phenol (PeCP) was bought from Anpel laboratory technologies (Shanghai) Inc. The quality control standard, 2,4,6- trichloro deuterated anisole (TCA-D5) was obtained from Dr. Ehrenstorfer. Methanol (HPLC grade) was purchased from Thermo Fisher Scientific (China) Co. Ltd. Analytical grade ethanol, potassium carbonate, and sodium chloride were produced by Fuchen (Tianjin) chemical reagent Co. Ltd. Analytical grade acetic anhydride was obtained from Sinopharm chemical reagent Co. Ltd. The red wine samples were purchased from the local market.
Solutions
The amount of 10.00 mg of each chemical standard was dissolved in a 10-mL volumetric flask with methanol to obtain 1.00 mg/mL stock solution as individual standard compound. The amount of 100 µL of each standard stock solution (except TCA-D5) was diluted with methanol to make 10 µg/mL mix-standard stock solution. The stock solutions were stored at − 20 ºC. Working solutions were diluted by methanol from the above stock solutions while being used. The amount of 1.38 g of potassium carbonate was put in 100 mL of water to make 0.1 mol/L potassium carbonate solution. Simulated wine was 12% ethanol solution.
Online Derivatization HS-SPME
The amount of 5.00 mL of wine sample was transferred into 20-mL head space crimp vials (Anple Laboratory technologies, Shanghai, China). A total of 1.0 g of sodium chloride, 100 µL of 100 µg/L TCA-D5 solution, 1.0 mL of 0.1 M potassium carbonate solution, and 100 µL of acetic anhydride and stirring bar were added into the head space vial. After sealing the vial by the cap with PTFE pad, the vial was stirring at 800 rpm and equilibrating at 60 ºC for 5 min, then 1 cm of 50/30 µm DVB/CAR/PDMS (Supelo, Bellefonte, PA, USA) was used to extract for 45 min, and the extracted samples were injected to GC at 270 ºC splitless for 5 min.
Instrumentation
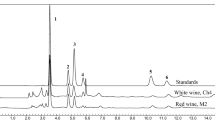
Agilent 7890A gas chromatograph-7000B triple quadrupole mass spectrometer (Agilent Technologies, Wilmington, DE, USA) were used for the quantitative analysis. HP-5 column (30.0 m × 0.25 mm × 0.25 µm, Agilent Technologies) was used for separation with helium (purity 99.9999%) as a carrier gas at a flow rate of 1.18 mL/min. GC oven temperature program was set as: the initial temperature was 50 ºC, kept for 1 min, ramped at 10 ºC/min to 200 ºC, then 40 ºC/min to 280 ºC, held for 2 min. The mass spectrometric detector (MSD) was operated as electron impact mode at 70 eV. The temperatures of the MSD transfer line, MSD source, and quadrupole were set at 270, 230, and 150 ºC respectively. Multiple reaction mode (MRM) was selected with 8 min solvent delay, two MS/MS transitions were selected, and optimized for each target chemicals according to the dependence of sensitivity and selectivity with collision energy. The formula, retention time, qualitative transition, quantitative transition, and their collision energy of each target compound were listed in Table 1. The total ion chromatography (TIC) and extracted ion chromatography (EIC) of each target compounds and isotopic control were shown in Figs. 1 and 2 respectively.

The total ionic chromatography of nine target compounds and isotopic control

The extracted ion chromatography of nine target compounds and isotopic control
Water purified by a Milli-Q water purification system (Millipore, Bedford, MA, USA) was used to prepare aqueous solutions. Magnetic stirrer (Anple, Shanghai, China), analytical balance (Mettler-Toledo, Switerland), MX-S vertex (Scilogex, USA), and KQ-600B Ultrasonic cleaner (Kunshan, Jiangsu, China) were also used during sample preparation.
Results and Discussion
Online Derivatization
Derivatization of MHPs is necessary because of their high polarity and low volatility; otherwise, the extraction efficiency of SPME and the separation efficiency of GC could not be satisfied for the determination of MHPs at trace concentration. Acetic anhydride is chosen as derivative reagent, MHPs will be transferred to corresponding acetate esters quickly and efficiently, thus can be detected as their acetate forms in the following MS analysis. MHAs will keep as initial structures under this condition. The usage amount of acetic anhydride was investigated at 0, 50, 100, and 200 µL. Figure 3 shows the usage amount of acetic anhydride vs target signal abundance.100 µL of acetic anhydride was selected as optimized derivatization amount.

Optimization of the derivatization MHPs with acetic anhydride
Optimization of HS-SPME
Generally, the extraction efficiency is strongly affected by some experimental parameters, such as the ionic strength, equilibrium time, extraction temperature, and extraction time. These parameters were optimized by using wine sample spiked with 100 µg/L standard mixture according to the corresponding GC–MS/MS abundance of different target compounds.
The addition of salt could improve the ionic strength of the matrix, assist the targets to escape from the matrix, increase their accumulation in the head space, and enhance the fiber absorption. However, over-addition of salt would increase the matrix viscosity, which might affect the targets escape from the matrix and reduce the extraction efficiency. Figure 4a is the summary of the optimization process of salt amount, 0, 0.05, 0.10, 0.25, 0.50, 1.00, and 2.00 g of sodium chloride were added to 5 mL of red wine, respectively. For most of the target compounds, the detection abundance reached maximum at 1.00 g of salt, so it was selected as optimized salt amount.

Optimization of the extraction process. a The effect of ionic strength; b the effect of equilibrium time; c the effect of extraction temperature; d the effect of extraction time
The extraction efficiency may also be influenced by the extraction condition since the physical properties of the target compounds (such as boiling point, polarity) are different. To obtain good extraction efficiency, the extraction parameters including the equilibrium time, extraction temperature, and extraction time were systematically studied as well. For equilibrium time, 0, 5, 10, 15, 20, and 30 min were studied, the results were summarized in Fig. 4 b. It showed there is no obviously difference among 5, 10, and 15 min of equilibrium time. In order to save time for sample preparation, 5 min of equilibrium time was chosen. The effect of the extraction temperature was inspected at 25, 30, 40, 50, 60, 70, 80, and 90 ºC, and the results were shown in Fig. 4 c. The abundance of all target compounds extracted first increases and then reaches the maximum value with the extraction temperature increasing. Although the temperatures to reach the maximum of abundance for different target compounds are different, 60 ºC was the compromise choice as optimized extraction temperature for all compounds. For the extraction time, 5, 10, 15, 20, 30, 45, and 60 min were examined respectively. The abundance of most of the targets reached maximum at 45 min of extraction time, except slight increment of 2,3,4,6-TeCA, 2,3,4,5-TeCA, PeCA, and PeCP with longer extraction time. Figure 4 d showed longer extraction time could increase the extraction efficiency, however, it would be time consuming for whole method operation.
Finally, the optimized SPME condition was selected as 5.00 mL of test sample, 1.0 g of sodium chloride, 5 min of equilibrium time, and extraction at 60 ºC for 45 min.
Standard Curves
In the previous reports, the simulated wine (12% ethanol-aqueous solution – v/v) was used to replace the matrix to construct the external calibration curves (Insa et al. 2004; ISO-20752 2014). We also investigated and compared the detection of the spiked 100 ng/L of the nine target compounds in simulated wine and red wine, respectively. There is no significant difference between them (Fig. 5). The usage of simulated wine instead of red wine could exclude the potential issue caused by the existing targets in the matrix-match calibration curve, especially for the determination of the commercial samples.

The comparison of 100 ng/L standard mixture in simulated wine and red wine
Linearity, LODs, and LOQs
The linear ranges, linear equations, correlation coefficient, LODs, and LOQs of the nine target compounds were listed in Table 2.
The linearity of the nine target chemicals was investigated under the optimal condition at 7 spiked concentration levels (10, 30, 100, 300, 1000, 3000, and 10,000 ng/L) with 5 duplicates. The external calibration curves were built by using the simulated wine. There is slightly difference of linearity ranges among these target compounds.
The LOQs of the nine target compounds were determined based on two criteria, one is the signal noise ratio greater than 10, and the other is that it must be included in the calibration curves. So the LOQs for all compounds were selected as the lowest point of the calibration curves: The LOQs of five MHAs were 10 ng/L, and the LOQs of MHPs were 30 ng/L (TCP and 2,3,4,6-TeCP) or 100 ng/L (TBP and PeCP). Although some compounds could be detected at much lower concentration levels, the LODs of these nine compounds were chosen as 1/3 of LOQs.
Recovery and Repeatability
After the calibration curves were built, the spiked recovery and repeatability— the important indexes to evaluate the developed method were investigated at three concentration levels (LOQs, 5*LOQs and 10*LOQs) in red wine, respectively. Six replicates were carried out at each concentration level. The recovery yields were the average of six replicates. Table 3 summarized the recovery yields and relative standard deviations (RSDs) of each target compound at different spiked levels. All of recovery yields were between 70 and 120%, which was satisfied the requirement of method development. The lowest recovery was 74.5%—TBA spiked at 50 ng/L, and the highest one was 119.5%—2,3,4,6-TeCA spiked at 10 ng/L. All of the RSDs were less than 20%, which could demonstrate the good repeatability of this proposed method. Based on the good recovery and repeatability mentioned above, this proposed method could be used to monitor the MHAs and MHPs in wine.
Validation with real samples
The developed method was validated with 16 commercial red wine samples which were purchased from the local markets. The original producers of these 16 wines are located in 5 countries, China, Chile, France, Spain, and Australia. Each sample was tested twice, the results were summarized in Table 4, and TCA, TCP, 2,3,4,6-TeCA, 2,3,4,5-TeCA, and PeCA were detected in 15 samples. In these commercial samples, TCP was detected most frequently; three wines have TCP concentration below LOQs (but detectable) and twelve wines have TCP concentration greater than LOQs, with the range from 52.3 to 2829 ng/L. In 6 commercial wine samples, TCA was detected, 5 of them with concentration below LOQs and the other one with concentration higher than LOQs as 75.6 ng/L. 2,3,4,6-TeCA, 2,3,4,5-TeCA, and PeCA were detected in 8, 3, and 4 out of 16 commercial samples respectively, with the concentrations below their LOQs.
Conclusions
Based on the powerful GC–MS/MS platform, through online derivatization of MHPs with acetic anhydride and head space solid phase microextraction, a quantitative detection method was developed to determine nine of MHAs and MHPs in wine. By optimizing the derivatization and extraction parameters, external calibration curves for each MHAs and MHPs were built by using simulated wine. Then the proposed method was validated by inspecting 16 commercial wine samples (produced in five countries) from local supermarket. Overall, four kinds of MHAs and one kind of MHPs were detected in 15 samples. The concentration of TCA detected in one sample and TCP detected in twelve samples are higher than their corresponding LOQs. 2,3,4,6-TeCA, 2,3,4,5-TeCA, and PeCA were also detected, but their concentrations are lower than their LOQs. Although TCP has less impact on the flavors of wine, its carcinogenicity should be concerned. This method could be potentially used as an inspection method for regulative organization to investigate MHAs and MHPs contents in wines in the future. Moreover, it also could be applied to determine the contents of MHAs and MHPs in drinking water, ground surface water, or other drinks when contamination happens.
Data Availability
All data generated or analyzed during this work are included in this published article.
References
Alvarez-Rodriguez ML, Lopez-Ocana L, Lopez-Coronado JM et al (2003) Cork taint of wines: role of the filamentous fungi isolated from cork in the formation of 2,4,6-trichloroanisole by o methylation of 2,4,6-trichlorophenol. Appl Environ Microb 68:5860–5869
Bertelli AAE (2007) Wine, research and cardiovascular disease: Instructions for use. Atherosclerosis 195:242–247
Boutou S, Chatonnet P (2007) Rapid headspace solid-phase microextraction/gas chromatographic/mass spectrometric assay for the quantitative determination of some of the main odorants causing off-flavours in wine. J Chromatogr A 1141:1–9
Campillo N, Viñas P, Cacho JI et al (2010) Evaluation of dispersive liquid–liquid microextraction for the simultaneous determination of chlorophenols and haloanisoles in wines and cork stoppers using gas chromatography–mass spectrometry. J Chromatogr A 1217:7323–7330
Chen C, Yu W, Wang W (2010) Red wine may be used in the therapy of myocarditis. J Cell Biochem 111:808–810
Coque JJR, Alvarez-Rodriguez ML, Larriba G (2003) Characterization of an inducible chlorophenol O-methyltransferase from Trichoderma longibrachiatum involved in the formation of chloroanisoles and determination of its role in cork taint of wines. Appl Environ Microb 69:5089–5095
Ezquerro Ó, Garrido-López Á, Tena MT (2006) Determination of 2,4,6-trichloroanisole and guaiacol in cork stoppers by pressurised fluid extraction and gas chromatography–mass spectrometry. J Chrom A 1102:18–24
Finley JW (2018) Beer and wine. Principles and Food Chemistry, 483–510 Springer International Publishing
Fontana AR (2012) Analytical methods for determination of cork-taint compounds in wine. TrAC-Trends Anal Chem 37:135–147
Hjelmeland AK, Collins TS, Miles JL et al (2012) High-throughput, sub ng/L analysis of haloanisoles in wines using HS-SPME with GC-Triple quadrupole MS. Am J Enol Vitic 63:494–499
IARC Working Group (2016) Pentachlorophenol and some related compounds. IARC Monogr Eval Carcinog Risk Chem Hum 117:4–11
Insa S, Salvadó V, Anticó E (2004) Development of solid-phase extraction and solid-phase microextraction methods for the determination of chlorophenols in cork macerate and wine samples. J Chromatogr A 1047:15–20
ISO-20752 International Standard (2014) Cork stopper – determination of releasable 2,4,6-trichloroanisol (TCA)
Lizarraga E, Irigoyen Á, Belsue V et al (2004) Determination of chloroanisole compounds in red wine by headspace solid-phase microextraction and gas chromatography–mass spectrometry. J Chromatogr A 1052:145–149
Martínez-Uruñuela A, Rodríguez I, Cela R et al (2005) Development of a solid-phase extraction method for the simultaneous determination of chloroanisoles and chlorophenols in red wine using gas chromatography–tandem mass spectrometry. Anal Chim Acta 549:117–123
Pizarro C, Pérez-del-Notario N, González-Sáiz JM (2007) Optimisation of a headspace solid-phase microextraction with on-fiber derivatisation method for the direct determination of haloanisoles and halophenols in wine. J Chromatogr A 1143:26–35
Pollnitz AP, Pardon KH, Liacopoulos D et al (1996) The analysis of 2,4,6-trichloroanisole and other chloroanisoles in tainted wines and corks. Aust J Grape Wine R 2:184–190
Requena T, Monagas M, Pozo-Bayón MA et al (2010) Perspectives of the potential implications of wine polyphenols on human oral and gut microbiota. Trends Food Sci Tech 21:332–344
Riu M, Mestres M, Busto O et al (2002) Determination of 2,4,6-trichloroanisole in wines by headspace solid-phase microextraction and gas chromatography-electron-capture detection. J Chromatogr A 977:1–8
Sefton MA, Simpson RF (2005) Compounds causing cork taint and the factors affecting their transfer from natural cork closures to wine - a review. Aust J Grape Wine R 11:226–240
Simpson RF, Sefton MA (2007) Origin and fate of 2,4,6-trichloroanisole in cork bark and wine corks. Aust J Grape Wine R 13:106–116
Taylor MK, Young TM, Butzke CE et al (2000) Supercritical Fluid Extraction of 2,4,6-Trichloroanisole from Cork Stoppers. J Agri Food Chem 48:2208–2211
Urquiaga I, Strobel P, Perez D et al (2010) Mediterranean diet and red wine protect against oxidative damage in young volunteers. Atherosclerosis 211:694–699
Vlachos P, Kampioti P, Kornaros M, Lyberatos G (2007) Matrix effect during the application of a rapid method using HS-SPME followed by GC–ECD for the analysis of 2,4,6-TCA in wine and cork soaks. Food Chem 105:681–690
Waterhouse AL, Sacks GL, Jeffery DW (2016) Understanding wine chemistry. John Wiley & Sons, Ltd., West Sussex, UK
Zalacain A, Alonso GL, Lorenzo C et al (2004) Stir bar sorptive extraction for the analysis of wine cork taint. J Chromatogr A 1033:173–178
Zhu BZ, Shen C, Gao HY et al (2017) Intrinsic chemiluminescence production from the degradation of haloaromaticpollutants during environmentally-friendly advance. J Environ Sci 62:68–83
Funding
This work was supported by the finacial support of State Adminstration for Market Regulation, China.
Author information
Authors and Affiliations
Contributions
Jialin Zhang and Xinying Li: investigation, data curation; Ang zhang: methodology, supervision; Chunlin Fan: conceptualization, funding acquisition, project administration; Zongyan Cui: data analysis and curation; Zijuan Zhang: methodology, funding acquisition, writing—original draft, writing—review & editing. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Competing interests
The authors declare no competing interests.
Conflict of Interest
Jialin Zhang declares that she has no conflict of interest. Ang Zhang declares that he has no conflict of interest. Chunlin Fan declares that he has no conflict of interest. Xinying Li declares that she has no conflict of interest. Zongyan Cui declares that he has no conflict of interest. Zijuan Zhang declares that she has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, J., Zhang, A., Fan, C. et al. Determination of Multihalo- Phenols and Anisoles in Wine by Gas Chromatography Tandem Mass Spectrometry Through Online Derivatization and Head Space Solid Phase Microextraction. Food Anal. Methods 15, 3435–3443 (2022). https://doi.org/10.1007/s12161-022-02371-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-022-02371-7