
Abstract
As common toxigenic fungi genera, Aspergillus, Penicillium, and Fusarium, have attracted the attention of governments and people all over the world due to their human carcinogenicity, teratogenicity, and hepatotoxicity. Accordingly, effective and quantitative detection method for toxigenic fungi before mycotoxins produced should be established to ensure food safety. In this paper, two duplex-droplet digital PCR (ddPCR) were developed and optimized for toxigenic fungi. The detection limits of the target genes, including the AflR, Och, Pen, and Fus of toxigenic fungi, were 26 copies/reaction, 15 copies/reaction, 161 copies/reaction, and 29 copies/reaction, respectively. Notably, the detection limit of duplex ddPCR was three orders of magnitude higher than that of tradition real-time fluorescence PCR reaction. Moreover the efficiency and sensitivity of the established method were also higher than those of real-time fluorescence PCR. The linear quantitative range of copy number of AflR and Och genes in AflR/Och duplex system and Pen and Fus genes in Pen/Fus duplex system were both 2 × 10−7–2 × 10−3 ng/μL. From DNA extraction to target gene detection, time consume of 96 samples was within 6 h, achieving the purpose of high-throughput and rapid detection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Toxigenic fungi are a class of fungi that can produce stimulating metabolites mycotoxins under the appropriate environmental conditions (Suman 2021). Until now, approximately 400 mycotoxins have been identified (Ülger et al. 2020). Ingestion, inhalation, or skin contact mycotoxins, even trace levels, would result in acute clinical symptom or chronic toxicity to humans and animals (Gallo et al. 2015). Antibiotic treatments have little or no effect on the diseases they caused (Marin et al. 2013). The genera Aspergillus, Fusarium, and Penicillium are the major mycotoxin-producing fungi, and the most relevant mycotoxins found in foods and feeds are aflatoxins (AFs), ochratoxins A (OTA), deoxynivalenol (DON), zearalenone(ZEN), patulin, and fumonisins (Marin et al. 2013). According to the statistics of the United Nations Food and Agriculture Organization (FAO), about 25% of the world’s grain and oil were contaminated by mycotoxins every year (Eskola et al. 2019). Though the methods of chromatography coupled to different detectors (Santos Pereira et al. 2019) and immunoassay-based methods (Pereira et al. 2014), biosensors (Santos Pereira et al. 2019) have been developed to detect mycotoxins in foodstuffs/feedstuffs, the annual costs in association with mycotoxin contamination on foods and feeds are continuing rise in worldwide (Hassan and Zhou 2018). Because mycotoxins are stable in nature and difficult to remove after pollution, it is an important measure to ensure the safety of agricultural products and foods to find fungal contamination as early as possible. Many national and international public health agencies, such as the World Health Organization (WHO), the Food and Agriculture Organization (FAO), and the U.S. Food and Drug Administration (FDA), are concerned about the problem of mycotoxins and have proposed control strategies (Mitchell et al. 2016). However, they have not yet addressed the issue of detecting and controlling those fungi with early toxin-producing properties, thus avoiding the risk at the source.
Direct detection of toxigenic fungi can more directly reduce the contamination of foods and the risk to humans compared to detection of mycotoxins (Alshannaq and Yu 2017). Earlier studies have been conducted on the use of PCR technology to detect aflatoxin-producing fungi (Haas et al. 2013; Niessen 2007; Stakheev et al. 2011). Multiplex PCR was also used to detect ochratoxin and Fusarium mycotoxin fungi (Sudharsan et al. 2017). However, the number of individual toxin-producing fungi is small in the early stage of food contamination, and the process of fungal enrichment is not only tedious but also time-consuming. Moreover, insufficient of fungal DNA often does not reach the detection limits of ordinary PCR. However, it seems that conventional PCR cannot do more in terms of target gene quantification (Raguseo et al. 2021).
Droplet digital polymerase chain reaction (ddPCR) is a recently proposed method that aims to overcome some of the limitations of qPCR in terms of sensitivity and precision (Yu et al. 2021). As a third-generation PCR technology, precise quantification can be achieved by directly counting the number of positive microdroplets after PCR reactions. The principle of ddPCR is to create a PCR reaction system with a fluorescent probe in oil and dilute it to the single molecule level, producing approximately 20,000 droplets. The detection of positive droplets and negative droplets is performed, and finally, precise quantification can be achieved by directly counting the number of positive microdroplets based on the Poisson distribution (Yu et al. 2020). Studies have been conducted to apply ddPCR technology to detect pathogenic bacteria such as Vibrio parahaemolyticus (Lei et al. 2020) and enterohemorrhagic Escherichia coli (He et al. 2020) in foods. However, to our knowledge, no quantitative analysis of toxigenic fungi in foods has been performed using a ddPCR method.
Herein, the objective of this work is to establish, optimize, and verify the accurate identification and quantitative methods of toxigenic fungi based on ddPCR. Specific primers and probes for the single-copy nuclear gene were designed for the identification of fungal DNA. In addition, a duplex ddPCR detection system was designed and optimized based on the two fluorescence channels of ddPCR to achieve simultaneous detection of two different toxigenic fungi. Finally, according to the target DNA sequence, the market samples were accurately quantified by ddPCR method.
Materials and Methods
Sample Collection
Escherichia coli TOP10 strain (Ampicillin resistance) and genomic DNA of Aspergillus flavus, Aspergillus parasiticus, Aspergillus ochraceus, Aspergillus nomius, Penicillium, and Fusarium were stored in our lab; among them DNA of Aspergillus flavus, Aspergillus parasiticus, and Aspergillus nomius were from aflatoxigenic strains confirmed by LC–MS. The DNA of fungal strains mentioned above were diluted to 20 ng/mL and stored at − 20 °C until used. DNA of Aspergillus flavus NRRL3357, Aspergillus ochraceus CBS263.67, Penicillium CBS131811, and Fusarium NRRL53570 were collected from the School of Food Engineering and Biotechnology, Tianjin University of Science and Technology. Food samples which eaten frequently in our daily lives and are prone to mold were purchased from several local supermarkets and farmers’ markets in Beijing, China (Tab. S1).
Primers and Probes
Specific target gene sequences of aflatoxin-producing fungi, Aspergillus ochra, Penicillium, and Fusarium were identified through Genbank, and the sequence homology comparison was performed using NCBI BLASTN (Fig.S1). The Och1 primer–probe pair was drawn from previous study (Patiño et al. 2005). Other primers and probes are designed using DNAMAN and Oligo7.0 software. A total of 8 sets of primer–probe pairs were screened by real-time PCR (data not shown); four sets of them were confirmed (Table 1). The oligonucleotide sequence was synthesized by Sangon Biotech (Shanghai, China) Co., Ltd., and the 5′ end of the probe was labeled with 6-carboxyfluorescein (FAM) or VIC, and the 3′ end was labeled with Black Hole Quencher 1 (BHQ1).
Construction of Target Genes Recombinant Plasmids
Four PCR products containing target genes, using the primer pairs in Table 1, were cloned into the vector pUC57 (Takara, Japan) according to the instructions of the manufacturer. Plasmids were transformed into Escherichia coli TOP10 cells and were purified by using a QIAGEN plasmid purification kit (QIAGEN, Germany) according to the manufacturer’s instructions. The quality and quantity of the extracted DNA were determined by spectrophotometry (NanoDrop One, USA) and diluted to 20 ng/μL and stored at − 20 °C until used.
The Establishment of ddPCR Assay
ddPCR reaction was carried out with QX200 Droplet Digital PCR system (Bio-Rad, USA), according to the manufacturer’s instructions. In the establishment of ddPCR assay, three ways to add restriction enzyme Hind III (R0104S, NEB, USA) to reaction mixture were test on plasmids in simplex assay (Table 2). Twenty microliters of mixtures contained 10 μL of 2 × ddPCR Supermix for probes (No dUTP) (Bio-Rad, USA), primers (10 μM) 1.8 μL each, probe (10 μM) 0.5 μL, DNA (20 ng/μL) 1 μL, and ddH2O 6.5 μL. A volume of 20 μL reaction mixture and 70 μL droplet generation oil for probes (Bio-Rad, USA) were used to generate droplets in an 8-channel cartridge with Bio-Rad-automated droplet generator. Then the generated emulsion droplets were transferred to a 96-well PCR plate using a RAININ electronic pipette set to 40 µL and sealed with foil in a PX1™ PCR plate sealer and then transferred to a T100™ Thermal Cycler for PCR amplification. PCR amplification was initiated by 10 min denaturation at 95 °C followed by 40 cycles of 95 °C for 15 s, 60 °C for 1 min, and 98 °C for 10 min to inactivate the enzyme and then stored at 4 °C. After amplification, PCR plate was transferred to the droplet reader, and data were analyzed by QuantaSoft™ software (version 1.6.6, Bio-Rad). A fluorescence amplitude threshold line was set up manually to distinguish between positive and negative droplets.
Specific of Droplet Digital PCR
The specificity of the primer pairs and probes were confirmed by simplex droplet digital PCR. Four constructed plasmids as mentioned above were tested and evaluated, and ddH2O was used as blank control. The optimized PCR reactions and parameters were used as described in section of the establishment of ddPCR assay.
Screening and Optimization of Duplex ddPCR Assay
Based on the FAM/VIC (HEX) dual-channel detection of Bio-Rad droplet digital PCR, two duplex ddPCR systems of aflatoxigenic fungi Aspergillus/Ochraceus (AflR/Och reaction system) and Penicillium/Fusarium (Pen/Fus reaction system) were established and optimized. For AflR/Och reaction system, DNA template mixtures were made by mixing 1μL pUC57-AflR (20 ng/μL) with 1μL pUC57-Och (20 ng/μL); primer–probe premix a was made by mixing AflR2 primers (10 μM) 1.8 μL each and AflR2 probe (10 μM) 0.5 μL, and primer–probe premix b was made by mixing Och2 primers (10 μM) 1.8 μL each and Och2 probe (10 μM) 0.5 μL; then five premix a/premix b ratio (3:1, 2:1, 1:1, 1:2, and 1:3) were tested and screened. The Pen/Fus reaction system were prepared and tested in a similar way (in this system: premix a was mixture of Pen primers and probe, premix b was mixture of Fus primers and probe). Duplex ddPCR reaction mixtures contained 10 μL of 2 × ddPCR Supermix for probes (No dUTP) (Bio-Rad, USA), different ratio primers-probe premix (10 μM) 4.1 μL, mixed template DNA (20 ng/μL) 1 μL, Hind III (20,000 units/mL) 0.2 μL, and ddH2O 4.7 μL. Five gradient annealing temperatures 60 ℃, 61 ℃, 62 ℃, 63 ℃, and 64 ℃ were set to select the best annealing temperature for the duplex ddPCR assay. The experimental data of the duplex system established by adjusting the proportional addition of primers and probes were analyzed by clustering of droplet fluorescence values, and the standard deviation of positive droplet fluorescence values was calculated using SPSS software.
Limit of Detection and the Linear Quantitative Equations of Duplex ddPCR System
Before the limit detection of the duplex ddPCR methods, the four plasmid DNA templates were mixed according to the ratio in the AflR/Och and Pen/Fus system to make a concentration of 20 ng/µL. Then the DNA were diluted into 10 gradients of 100–109 and amplified in optimized duplex ddPCR reaction system. Experiments were set up in triplicate, and repeated experiments were carried out among the experimenters. The linear quantitative equations of duplex ddPCR method were also established according to Ren et al (2017).
Selection of DNA Extraction Methods and Analysis of Actual Samples
The quality of DNA has an enormous impact on detection results. Before extraction, all food samples were grind into a fine power by liquid nitrogen; subsequently, one gram of grinded sample was placed in a 5 mL Axygen tube. For optimum method selection of DNA extraction, three DNA extraction methods were used. Method 1 was a cetyltrimethylammonium bromide (CTAB) method as described by Li et al. (2017), and method 2 and method 3 were commercial kit-based methods with NucleoSpin®Food kit (MP Biomedicals, USA) and FastDNA™ SPIN Kit (Macherey–Nagel, Germany), respectively, used according to the instruction.
To verify the accuracy of the developed double-stranded ddPCR method for target gene quantification, genomic DNA of previously mentioned fungal strains with known concentrations was examined. To further demonstrate the accuracy of the method for the actual food samples, eight food samples including grape, steamed bread, toast, sandwiched bread, northeast rice, vitamin compound nutritious rice noodles, mineral compound nutritious rice noodles, and calcium compound nutritious rice noodles were also detected. DNA extraction of food samples were performed and amplified by optimized duplex ddPCR systems as above-mentioned.
Results
Specificity of Primer Pairs and Probes
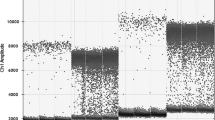
The specificity of the four primer pairs and probes were tested with the four recombinant plasmids (pUC57-AflR, pUC57-Och, pUC57-Pen, pUC57-Fus). The results showed that ddPCR amplification of the four plasmid DNA with each set of primer pairs and probe produced a specific signal of expected (Fig. 1). In short, when using AflR2 primer pairs and probe, specific signals were the recombinant plasmid pUC57-AflR, the other plasmid DNA samples as well as blank control were all negative, and primer pairs and probes of Och2, Pen, and Fus were similar. And the number of droplets generated in each sample was more than 12,000, satisfying the Poisson distribution calculation. Meanwhile, the primers did not cross-react between different samples, indicating that the digital PCR system has good specificity of primers and probes and can be used for subsequent studies.

The specificity of the primer pairs and probes in ddPCR assay. The specific droplets were obtained from four plasmid DNA (pUC57-AflR, pUC57-Och, pUC57-Pen, pUC57-Fus) with different primer pairs and probe: a four recombinant plasmids amplification with AflR2 primer pairs and probe; b four recombinant plasmids amplification with Och primer pairs and probe; c four recombinant plasmids amplification with Pen primer pairs and probe; d four recombinant plasmids amplification with Fus primer pairs and probe.
Establishment of ddPCR Assay
At present study, the ddPCR assay was set up to identified the ways to add restriction enzyme. Figure S2 showed that droplet separation was visibly best in a system than in b and c. And the standard deviation of positive versus negative droplets was also calculated; the smallest value was observed in a system (Fig. 2). Therefore system a was chosen and used in subsequent ddPCR assay.

Comparison of the mean difference and standard deviation of fluorescence of positive/negative droplets of ddPCR systems in different ways to add restriction enzyme Hind III. a Hind III was added in the reaction mixtures; b Hind III and CutSmart® Buffer were added in the reaction mixtures; c plasmid DNA was digested with Hind III and purified before ddPCR and then was added in the reaction mixtures
Optimization of Duplex ddPCR System
The two duplex ddPCR initially optimized the ratio of premix a to premix b in the reaction systems. In AflR/Och duplex system, when the ratio of premix a to premix b was 1:3, the positive droplets were better clustered at this time (Fig. 3a), and the value of the standard deviations of the AflR/Och duplex system was the smallest (Fig. 3c). In Pen/Fus duplex system, the positive droplets clustered better when the ratio of premix a to premix b was 3:1 (Fig. 3b), and also the value of the standard deviations was the smallest (Fig. 3d). Therefore, the ratio of 1:3 and 3:1 of premix a to b in AflR/Och and Pen/Fus, respectively, were identified as the most appropriate reaction conditions, since the fluorescence reporting values of the two duplex reaction systems were high and with better separate areas between the negative and positive microdroplets cluster.

Optimization of duplex ddPCR primer pairs and probes. a Two-dimensional plots of fluorescence amplitudes with the ratio of premix a to premix b were 1:3 in AflR/Och duplex system; b two-dimensional plots of fluorescence amplitudes with the ratio of premix a to premix b were 3:1 in Pen/Fus duplex system; c comparison of the mean difference and standard deviation of fluorescence of positive/negative droplets in AflR/Och ddPCR system of different ratio of premix a to premix b; d comparison of the mean difference and standard deviation of fluorescence of positive/negative droplets in Pen/Fus ddPCR system of different ratio of premix a to premix b. Blue and orange dots represent the positive droplets, and black and green dots represent the negative droplets
According to the ddPCR application guidelines, annealing temperature is one of the key parameters affecting specificity. In order to determine the optimum annealing temperature of ddPCR duplex system, five different temperatures (60℃, 61℃, 62℃, 63℃, and 64℃) were designed and compared. The best separation between positive and negative droplets for the targets in two duplex systems were observed at the lowest tested temperatures, and therefore, 60℃ and 61℃ were identified as the most appropriate annealing temperatures in AflR/Och system (Fig. 4a, b) and Pen/Fus system (Fig. 4c, d), respectively, since they presented slightly less droplet rain between positive and negative droplets compared to the other tested temperatures. Similar results were obtained using the standard deviation calculated and analyzed; there were minimum values at 60℃ in AflR/Och system (Fig. 4e) and 61℃ Pen/Fus system (Fig. 4f), respectively.

Optimization of duplex ddPCR annealing temperatures. a AflR in AflR/Och duplex-system, at annealing temperatures 60 °C, 61 °C, 62 °C, 63 °C, and 64 °C; b Och in AflR/Och duplex system, at annealing temperatures 60 °C, 61 °C, 62 °C, 63 °C, and 64 °C; c Pen in Pen/Fus duplex-system, at annealing temperatures 60 °C, 61 °C, 62 °C, 63 °C, and 64 °C; d Fus in Pen/Fus duplex-system, at annealing temperatures 60 °C, 61 °C, 62 °C, 63 °C, and 64 °C; e comparison of the mean difference and standard deviation of fluorescence of positive/negative droplets in AflR/Och duplex ddPCR system of five annealing temperatures; f comparison of the mean difference and standard deviation of fluorescence of positive/negative droplets in Pen/Fus duplex ddPCR system of five annealing temperatures. Blue and green dots represent the positive droplets, and black dots represent the negative droplets
LOD and Quantitative Linear Range of Duplex ddPCR
Ten-fold serial mixed plasmid dilutions were tested and used to construct the linear quantitative curve by putting the plasmid DNA concentration logarithm against the measures copy number logarithm. The LODs of AflR, Och, Pen, and Fus genes were 26 copies/reaction, 15 copies/reaction, 161 copies/reaction, and 29 copies/reaction, respectively, calculated by Poisson distribution and eliminating blank aerosol contamination. And after more than two experiments and the replacement of experimental personnel, the experimental results are consistent with expectations, and there is a good repeatability and reproducibility of the experiment (results not show).
The linear quantification range in this study refers to the concentration range in which the assay can stably quantify target genes for toxigenic fungi and is an evaluation of the detection ability of the system. By linear fit analysis, the quantitative ranges of AflR, Och, Pen, and Fus gene copy numbers in both AflR/Och and Pen/Fus duplex systems were 2 × 10−7 to 2 × 10−3 ng/μL, and the linear quantitative equations for the four genes were shown in Fig. 5.

Linear regression of the duplex ddPCR. a Copy numbers vs. plasmid DNA of AflR in AflR/Och duplex system; b copy numbers vs. plasmid DNA of Och in AflR/Och duplex system; c copy numbers vs. plasmid DNA of Pen in Pen/Fus duplex system; d copy numbers vs. plasmid DNA of Fus in Pen/Fus duplex system
Selection of Extraction Methods and Detection of DNA in Actual Samples
Eight samples, Jufeng grape, steamed bread, toast, sandwiched bread, northeastern rice, vitamin compound nutritious rice noodles, mineral compound nutritious rice noodles, and calcium compound nutritious rice noodles, were numbered as 1–8, respectively. The results were shown in Table S1, and the concentration of DNA in the table is the concentration extracted from 1-g food samples. The results showed that the extraction rate of DNA extracted by FastDNA™ SPIN Kit is the highest and the intra-group error is low. Sample 6, sample 7, and sample 8 are deeply processed foods, which may be the reason why the yield of DNA is generally low. Then the DNA of food samples extracted by three DNA extraction methods were added to the two established duplex ddPCR reaction systems for amplification. The amplification results of DNA extracted by FastDNA™ SPIN Kit are listed in Table 3, and the amplification results of the other two methods are shown in Table S2 and Table S3. According to all the quantitative results, the DNA of food samples extracted by FastDNA™ SPIN Kit showed the highest detection rate of 40.63% of toxigenic fungal genes in two duplex ddPCR reaction systems.
Steamed breads and northeastern rice were the food products with the highest rate of detection of toxic fungi, with the detection of toxic fungal genes AFlR, Och, and Fus and Och, Pen, and Fus, respectively. Steamed breads had the highest AFlR gene copy number of 3,763 copies/μL and Fus gene copy number of 4,285 copies/μL; toast had the highest Och gene copy number of 2,930 copies/μL; Jufeng grape had the highest Pen gene copy number of 3,123 copies/μL.
To determine the accuracy of the duplex ddPCR methods, genomic DNA of fungal strains with known concentration was analyzed. Genomic DNA was extracted from each fungus three times, and three independent replicate assays were performed. The DNA copies were calculated according to the linear quantitative equations for the four genes as shown in Fig. 5. The amounts between 2 × 10−3 and ~ 2 × 10−7 ng DNA per μL were all quantified. The results showed that for AflR quantification, it was detected in Aspergillus flavus, Aspergillus parasiticus, and Aspergillus terreus. The deviation from the theoretical value was − 89.77 to 0.54 (copies/μL), and the relative standard deviation (RSD) was between 0.32 and 18.78% (Table 4), far less than 25%, following FAO Guidelines for identification and quantification based on specific DNA sequences (Commission 2010). Meanwhile, Och, Pen, and Fus were tested in Aspergillus ochraceus, Penicillium, and Fusarium, respectively. The relative standard deviations ranged from 0.38 to 15.64, which verified the accuracy of the established method.
Discussion
Mycotoxins are widely found in grains (Varzakas 2016), nuts (Rodrigues et al. 2012), fruits, soft drinks, alcoholic beverages (Alshannaq and Yu 2017; Welke 2019), and other agricultural products, which continue to have a great negative impact on food safety, human health, agricultural economy, and so on. The production of mycotoxins can occur at any stage of food production, which makes detection difficult because mycotoxins do not have characteristic odors and do not change the sensory properties of foods (Agriopoulou et al. 2020). Therefore, the accurate and quantitative detection of toxigenic fungi before the occurrence of mycotoxins is the most important part to control mycotoxin pollution. In earlier years, Gil-Serna et al. have described specific PCR methods based on multi-copy ITS regions to detect and identify major ochratoxigenic species of Aspergillus in foodstuffs (Gil-Serna et al. 2009). Similarly, Sardiñas et al. (2011) tested the PCR amplification ability of DNA obtained from 93 samples of paprika and pepper by using specific primers for 7 Aspergillus species. In some recent work, von Hertwig et al. proposed a real-time PCR method for the detection of Aspergillus niger/Aspergillus wedii (von Hertwig et al. 2018). But some mycotoxins are produced by more than one fungus, while others can produce more than one mycotoxin (Zain 2011). Based on this, multiplex PCR technique was applied to the detection of toxigenic fungi. “Multiplex PCR” (mPCR) refers to the simultaneous amplification of multiple targets in a single reaction by using more than one pair of primers. By targeting more than one gene at a time, additional information can be obtained from a single test (Hayat et al. 2012). Sim et al. established two multiplex PCR methods that can sensitively and specifically detect zearalenone-producing fungi in rice (Sim et al. 2018). The two mPCR methods show high sensitivity in detecting ZEA producing species, and the LOD is 1.25 pg/μL of genomic DNA. Rodríguez et al. established a method for quantitative production of aflatoxin-producing mold and ochratoxin A (OTA)-producing mold in foodstuffs based on TaqMan probe multiple real-time qPCR technology (Rodríguez et al. 2012). And the limit of detection was between 3 and 1 log cfu/g in all inoculated foods depending on the tested mycotoxin producer. In this work, on the basis of predecessors, two duplex droplet digital PCR systems were established to realize the absolute quantitative detection of four kinds of toxigenic fungi. Notably, addition of Hind III to the reaction could improve the aggregation of droplets, and the assay could be further optimized by regulating the premix a to b ratio in the two systems. Moreover the optimum annealing temperature was 60℃ and 61℃ for the AflR/Och and the Pen/Fus detection system, respectively. The detection limits of AflR, Och, Pen, and Fus are 26 copies/reaction, 15 copies/reaction, 161 copies/reaction, and 29 copies/reaction, respectively. The detection limits of duplex ddPCR were two to three orders of magnitude higher than those of our previously established qPCR method, while the detection limits for Aspergillus ochraceus, Penicillium, and Fusarium were 3.37 × 104, 1.91 × 104, 1.53 × 104, and 3.95 × 104 copies/reaction, respectively (Zhang et al. 2020). The linear quantitative range of copy number of AflR and Och genes in A/O duplex system is 2 × 10−7–2 × 10−3 ng/μL, and the linear quantitative range of copy number of Pen and Fus genes in P/F duplex system is 2 × 10−7–2 × 10−3 ng/μL. As shown in Fig. 5, when the amount of target DNA in the sample is higher than the linear quantification range, the DNA concentration in the reaction system is too high, and the recombinant plasmid molecules contained in a single droplet are greater than or equal to 1 during droplet formation, forming a supersaturated state that no longer conforms to the Poisson distribution. Therefore, when the DNA content is high, the copy number of the target gene remains essentially constant. When the DNA content in the sample is below the linear quantification range, the copy number obtained at this point is equal to that of the blank control and can be considered as not amplified.
PCR and qPCR methodologies have been widely used in the detection of toxin-producing fungi. The existence of a variety of substances that inhibit PCR amplification in foods and the environment (Chen et al. 2017). This can lead to a decrease in sensitivity, leading to the emergence of false negative results. The ddPCR can solve part of this problem, because during the process, each template molecule is amplified in a separate reaction chamber, which is produced by droplets emulsified in oil or a microfluidic device (Hindson et al. 2011). The droplet has the same effect as a single test tube. The ddPCR system divides the DNA sample into millions of droplets to support PCR amplification of template molecules (Chen et al. 2017). In our results, positive droplets were clearly distinguished from negative droplets, and there were no false negatives in the ddPCR results. There was also no cross-reactivity between specific and heterogeneous primers and each sample (Fig. 1). The reason why the blank control produces very few signals is very likely to be the high sensitivity of digital PCR. The false positive results caused by aerosol pollution in the laboratory can be solved by the appropriate threshold line in the quantitative process.
In the existing research, ddPCR was applied to the identification of true properties of foods (Yu et al. 2021) and the detection of pathogenic bacteria in foodstuffs (He et al. 2020; Lei et al. 2020). As far as we know, this is the first time that ddPCR methodology has been applied to the detection of toxigenic fungi. High-quality specific primers and probes were designed, multiple ddPCR reaction conditions were established and optimized, and the detection sensitivity was improved. The toxigenic fungi can be detected accurately and quantitatively during the latent period.
In addition, we used the constructed two duplex ddPCR systems to detect the DNA of 6 kinds of molds (Aspergillus flavus, Aspergillus parasiticus, Aspergillus ochraceus, Aspergillus nomius, Penicillium, Fusarium) to repeatedly verify the reliability of the system. Among them, Aspergillus parasiticus and Aspergillus nomius were not detected in our system, which proved the excellent specificity of the two duplex ddPCR (data not shown in the results).
Conclusions
In summary, a method for the detection of potentially toxigenic fungi in foods by two duplex ddPCR systems was established. This method improves the sensitivity for the detection of toxin-producing fungi, has good repeatability, and can complete the detection of no more than 96 samples within 6 h. It meets the requirements of high-throughput and rapid detection. This work provides technical support for predicting the toxin-producing performance of latent toxigenic fungi and pre-intervention of toxin hazards in the early stage of toxin formation and provides a reference for effective prevention and control of toxin-contaminated food and ensuring food safety.
Data Availability
The datasets supporting the conclusions of this article are included within the article and its additional files.
References
Agriopoulou S, Stamatelopoulou E, and Varzakas T (2020) Advances in occurrence, importance, and mycotoxin control strategies: prevention and detoxification in foods. Foods 9(2) 137 https://www.mdpi.com/2304-8158/9/2/137
Alshannaq A, and Yu JH (2017) Occurrence, toxicity, and analysis of major mycotoxins in food. Int J Environ Res Public Health 14(6) 632 https://www.mdpi.com/1660-4601/14/6/632
Chen J-Q, Healey S, Regan P, Laksanalamai P, Hu Z (2017) PCR-based methodologies for detection and characterization of Listeria monocytogenes and Listeria ivanovii in foods and environmental sources. Food Sci Human Wellness 6(2):39–59. https://doi.org/10.1016/j.fshw.2017.03.001
Commission CA (2010) CAC/GL 74-2010. Guidelines on performance criteria and validation of methods for detection, identification and quantification of specific DNA sequences and specific proteins in foods. Rome: Codex Alimentarius Commission
Eskola M, Kos G, Elliott CT, Hajšlová J, and Mayar S (2019) Worldwide contamination of food-crops with mycotoxins: validity of the widely cited 'FAO estimate' of 25%. Critical Rev Food Sci Nutr 16(60):2773–2789. https://doi.org/10.1080/10408398.2019.1658570
Gallo A, Giuberti G, Frisvad JC, Bertuzzi T, Nielsen KF (2015) Review on mycotoxin issues in ruminants: occurrence in forages, effects of mycotoxin ingestion on health status and animal performance and practical strategies to counteract their negative effects. Toxins 7:3057–3111. https://doi.org/10.3390/toxins7083057
Gil-Serna J, Vázquez C, Sardiñas N, González-Jaén MT, Patiño B (2009) Discrimination of the main ochratoxin A-producing species in Aspergillus section Circumdati by specific PCR assays. Int J Food Microbiol 136(1):83–87. https://doi.org/10.1016/j.ijfoodmicro.2009.09.018
Haas D, Pfeifer B, Reiterich C, Partenheimer R, Reck B, Buzina W (2013) Identification and quantification of fungi and mycotoxins from Pu-erh tea. Int J Food Microbiol 166(2):316–322. https://doi.org/10.1016/j.ijfoodmicro.2013.07.024
Hassan YI, and Zhou T (2018) Promising detoxification strategies to mitigate mycotoxins in food and feed. Toxins, 10(3), 116 https://www.mdpi.com/2072-6651/10/3/116
Hayat A, Paniel N, Rhouati A, Marty J-L, Barthelmebs L (2012) Recent advances in ochratoxin A-producing fungi detection based on PCR methods and ochratoxin A analysis in food matrices. Food Control 26(2):401–415. https://doi.org/10.1016/j.foodcont.2012.01.060
He L, Simpson DJ, Gänzle MG (2020) Detection of enterohaemorrhagic Escherichia coli in food by droplet digital PCR to detect simultaneous virulence factors in a single genome. Food Microbiol 90:103466. https://doi.org/10.1016/j.fm.2020.103466
von Hertwig AM, Sant'Ana AS, Sartori D, da Silva JJ, Nascimento MS, Iamanaka BT, . . . Taniwaki MH (2018) Real-time PCR-based method for rapid detection of Aspergillus niger and Aspergillus welwitschiae isolated from coffee. J Microbiol Methods 148 87-92 https://doi.org/10.1016/j.mimet.2018.03.010
Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, . . . Legler TC (2011) High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal chem 83 (22), 8604-8610. https://doi.org/10.1021/ac202028g
Lei S, Gu X, Zhong Q, Duan L, Zhou A (2020) Absolute quantification of Vibrio parahaemolyticus by multiplex droplet digital PCR for simultaneous detection of tlh, tdh and ureR based on single intact cell. Food Control 114:107207. https://doi.org/10.1016/j.foodcont.2020.107207
Li H, Fu T, Yun Z, Lv T, Yuan L, Xu B (2017) Effect comparison of five methods to extract fungal genomic DNA as PCR templates. Chin Agric Sci Bull. https://doi.org/10.1016/j.fct.2013.07.047
Marin S, Ramos AJ, Cano-Sancho G, Sanchis V (2013) Mycotoxins: occurrence, toxicology, and exposure assessment. Food Chem Toxicol 60:218–237. https://doi.org/10.1016/j.fct.2013.07.047
Mitchell NJ, Bowers E, Hurburgh C, Wu F (2016) Potential economic losses to the US corn industry from aflatoxin contamination. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 33(3):540–550. https://doi.org/10.1080/19440049.2016.1138545
Niessen L (2007) PCR-based diagnosis and quantification of mycotoxin producing fungi. Int J Food Microbiol 119(1):38–46. https://doi.org/10.1016/j.ijfoodmicro.2007.07.023
Patiño B, González-Salgado A, González-Jaén MT, Vázquez C (2005) PCR detection assays for the ochratoxin-producing Aspergillus carbonarius and Aspergillus ochraceus species. Int J Food Microbiol 104(2):207–214. https://doi.org/10.1016/j.ijfoodmicro.2005.02.011
Pereira VL, Fernandes JO, Cunha SC (2014) Mycotoxins in cereals and related foodstuffs: a review on occurrence and recent methods of analysis. Trends Food Sci Technol 36(2):96–136. https://doi.org/10.1016/j.tifs.2014.01.005
Raguseo C, Gerin D, Pollastro S, Rotolo C, Rotondo PR, Faretra F, De MiccolisAngelini RM (2021) A duplex-droplet digital PCR assay for simultaneous quantitative detection of Monilinia fructicola and Monilinia laxa on Stone Fruits. Front Microbiol 12:747560–747560. https://doi.org/10.3389/fmicb.2021.747560
Ren J, Deng T, Huang W, Chen Y, Ge Y (2017) A digital PCR method for identifying and quantifying adulteration of meat species in raw and processed food. PLoS ONE 12(3):e0173567–e0173567. https://doi.org/10.1371/journal.pone.0173567
Rodrigues P, Venâncio A, Lima N (2012) Mycobiota and mycotoxins of almonds and chestnuts with special reference to aflatoxins. Food Res Int 48(1):76–90. https://doi.org/10.1016/j.foodres.2012.02.007
Rodríguez A, Rodríguez M, Andrade MJ, Córdoba JJ (2012) Development of a multiplex real-time PCR to quantify aflatoxin, ochratoxin A and patulin producing molds in foods. Int J Food Microbiol 155(1):10–18. https://doi.org/10.1016/j.ijfoodmicro.2012.01.007
Santos Pereira C, Cunha SC, Fernandes JO (2019) Prevalent mycotoxins in animal feed: occurrence and analytical methods. Toxins 11(5):290. https://doi.org/10.3390/toxins11050290
Sardiñas N, Gil-Serna J, Santos L, Ramos AJ, González-Jaén MT, Patiño B, Vázquez C (2011) Detection of potentially mycotoxigenic Aspergillus species in Capsicum powder by a highly sensitive PCR-based detection method. Food Control 22(8):1363–1366. https://doi.org/10.1016/j.foodcont.2011.02.013
Sim JH, Tian F, Jung SY, Auh J-H, Chun HS (2018) Multiplex polymerase chain reaction assays for the detection of the zearalenone chemotype of Fusarium species in white and brown rice. Int J Food Microbiol 269:120–127. https://doi.org/10.1016/j.ijfoodmicro.2018.02.003
Stakheev AA, Ryazantsev DY, Gagkaeva TY, Zavriev SK (2011) PCR detection of Fusarium fungi with similar profiles of the produced mycotoxins. Food Control 22(3):462–468. https://doi.org/10.1016/j.foodcont.2010.09.028
Sudharsan S, Malka B, Varda Z, Moshe K, Anatoly T, Elazar Q, Edward S (2017) Rapid detection and identification of mycotoxigenic fungi and mycotoxins in stored wheat grain. Toxins 9(10):302. https://doi.org/10.3390/toxins9100302
Suman M (2021) Last decade studies on mycotoxins’ fate during food processing: an overview. Curr Opin Food Sci 41:70–80. https://doi.org/10.1016/j.cofs.2021.02.015
Ülger TG, Uçar A, Çakıroğlu FP, Yilmaz S (2020) Genotoxic effects of mycotoxins. Toxicon 185:104–113. https://doi.org/10.1016/j.toxicon.2020.07.004
Varzakas T (2016) Quality and safety aspects of cereals (wheat) and their products. Crit Rev Food Sci Nutr 56(15):2495–2510. https://doi.org/10.1080/10408398.2013.866070
Welke JE (2019) Fungal and mycotoxin problems in grape juice and wine industries. Curr Opin Food Sci 29:7–13. https://doi.org/10.1016/j.cofs.2019.06.009
Yu N, Ren J, Huang W, Xing R, Chen Y (2021) An effective analytical droplet digital PCR approach for identification and quantification of fur-bearing animal meat in raw and processed food. Food Chem 10:129525. https://doi.org/10.1016/j.foodchem.2021.129525
Yu N, Han J, Deng T, Chen L, Zhang J, Xing R, . . . Chen Y (2020) A novel analytical droplet digital PCR method for identification and quantification of raw health food material powder from Panax notoginseng. Food Analytical Methods 1-9 https://doi.org/10.1007/s12161-020-01887-0
Zain ME (2011) Impact of mycotoxins on humans and animals. J Saudi Chem Soc 15(2):129–144. https://doi.org/10.1016/j.jscs.2010.06.006
Zhang Q, Zhao X, Chen Y, Liu B, Wang P (2020) [Establishment of rapid detection method for multiple real-time fluorescent PCR of toxin-producing fungi]. Wei Sheng Yan Jiu = J Hyg Res 49(6):881–888. https://doi.org/10.19813/j.cnki.weishengyanjiu.2020.06.002
Funding
This work was supported by the National Key Research and Development Program of China (Grant Number 2017YFC1601200).
Author information
Authors and Affiliations
Contributions
Ping Wang: formal analysis, writing—review and editing, investigation, and validation. Tianming Qu: methodology, formal analysis, writing—original draft. Xiaomei Zhao: data curation, methodology, supervision. Yiqiang Ge: methodology and formal analysis. Ying Chen: conceptualization, writing—review and editing, supervision, project administration, and funding acquisition.
Corresponding author
Ethics declarations
Ethics Approval
This article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable.
Conflict of Interest
Ping Wang declares that she has no conflict of interest. Tianming Qu declares that he has no conflict of interest. Xiaomei Zhao declares that she has no conflict of interest. Yiqiang Ge declares that he has no conflict of interest. Ying Chen declares that she has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, P., Qu, T., Zhao, X. et al. An Efficient Droplet Digital PCR Approach for Detection DNA at Low Concentrations of Toxigenic Fungi in Food Products. Food Anal. Methods 15, 1695–1706 (2022). https://doi.org/10.1007/s12161-022-02236-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-022-02236-z