
Abstract
This study reports an analytical method to simultaneously determine the residues of sulfonamides, dapsone, ormethoprim, and trimethoprim in fish and shrimp. The samples were extracted with acetonitrile, magnesium sulfate, and sodium acetate, followed by dispersive solid-phase extraction (C18) for cleanup. The target analytes were confirmed and quantified by liquid chromatography–tandem mass spectrometry. The developed method was validated according to the Codex guidelines (CAC/GL 71-2009). As a result, linearity was expressed between 0.25 and 200 μg kg−1 with a correlation coefficient > 0.98. The limits of quantitation were 0.01–2.8 μg kg−1 and the accuracy (expressed as average recovery) was 76.1–115%. The precision (expressed as the coefficient of variation) was <20%. The decision limits of sulfonamides, dapsone, ormethoprim, and trimethoprim were 94.2–114, 1.3–1.5, 105–111, and 53.3–54 μg kg−1 and detection capabilities of 102–129, 1.4–1.6, 109–120, and 55.6–58.2 μg kg−1, respectively. The proposed method was applied to the analysis of real samples (n = 54) obtained from domestic markets in Korea, resulting in a detection rate of 13% (seven samples). This method is applicable for the efficient determination of sulfonamides, dapsone, ormethoprim, and trimethoprim residues in fish and shrimp according to Korean MRL.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sulfonamides are widely used to treat animal infections and promote growth to support farm animal health (Song et al. 2017). Sulfonamides are one of the most popular antibiotics due to their low price, low toxicity, and excellent antibacterial activity against common bacterial infections (Baran et al. 2011). Trimethoprim, ormethoprim, and dapsone demonstrate antibacterial activities that are similar to sulfonamides, which are frequently used in combination for clinical treatment (Nicosia et al. 2014; Economou et al. 2012). In particular, antibiotic diaminopyrimidines (e.g., trimethoprim and ormethoprim) are often combined with sulfonamides, resulting in enhanced bactericidal efficacy (Serrano 2005; Reeves 2012). The uncontrolled use of these antibiotics and noncompliance with guidelines for withdrawal periods when treating animals raised to obtain food products endangers humans by contributing to the development of antibiotic resistance (Economou et al. 2012; McDonald et al. 2009; Hoff et al. 2015).
Regulatory authorities have established maximum residue limits (MRLs) for total sulfonamides in farmed aquatic animal products as 100 μg kg−1, and MRLs of 50 and 100 μg kg−1 for trimethoprim and ormethoprim, respectively (CAC 2017; EC European Commission 2009a, b; CAC 2015; USDA 2017). In compliance with MRL, several countries (Korea, Portugal, China, Greece, and Brazil) have managed to control the use of these antibiotics in food products (Song et al. 2017; Economou et al. 2012; Hoff et al. 2015; Freitas et al. 2014; Kang et al. 2019a; Kang et al. 2019b). In the 1960s, dapsone and sulfonamides were used as both antibacterial and anti-inflammatory agents (Zhu and Stiller 2001). However, dapsone has been banned for use in veterinary treatments for food producing animals based on genotoxicity studies (EC European Commission 2009b).
In order to ensure the stipulated residue levels, analytical methods with sufficient sensitivity and specificity to detect and quantify residue levels in food are needed. Several authors have developed analytical methods to determine the residues of sulfonamides. For instance, methods using liquid chromatography–tandem mass spectrometry (LC–MS/MS) to detect sulfonamides or sulfonamides with trimethoprim in fishery products (Freitas et al. 2014; Liu et al. 2014; Kang et al. 2018; Shin et al. 2018), ultra-high performance liquid chromatography (UHPLC) Q-Orbitrap to detect sulfonamides in salmon (Jia et al. 2018), LC–MS/MS for detection of sulfonamides in tilapia fillet (Nunes et al. 2018), and liquid chromatography quadrupole time-of-flight mass spectrometry (LC-Q TOF/MS) to analyze the residues of sulfonamides and trimethoprim in fish tissue (Li et al. 2016) have been developed. However, there are no reported multi-residue methods for sulfonamides combined with dapsone (its metabolite), ormethoprim, and trimethoprim in several matrices.
In this work, an analytical method was developed based on a modified QuEChERS (quick, easy, cheap, effective, rugged, and safe) method using dispersive solid-phase extraction (d-SPE) combined with LC–MS/MS. The proposed method was evaluated for three kinds of fish and shrimp matrices under remarkable validation criteria to facilitate a rapid, sensitive, and reliable method for the simultaneous determination of 24 antibiotics (21 kinds of sulfonamides, dapsone, ormethoprim, and trimethoprim). Moreover, the developed method was applied in real sample analysis to confirm the simultaneous determination of target compounds in fishery product samples purchased from a local Korean market. The developed method is the first report to demonstrate the screening and confirmation of multiple compound residues in fish and shrimp.
Materials and Methods
Chemicals and Materials
High purity (>90%) standards (sulfonamides, dapsone, ormethoprim, and trimethoprim) were purchased from Sigma-Aldrich (MO, USA), Dr. Ehrenstorfer (Augsburg, Germany), and US Pharmacopeia (MD, USA). These standards were dissolved in methanol to a concentration of 1000 μg mL−1 and stored at −10 °C. HPLC-grade methanol was purchased from Merck (Darmstadt, Germany); magnesium sulfate, sodium acetate, and formic acid were purchased from Sigma-Aldrich (MO, USA); octadecylsilane (C18) was purchased from Waters (MA, USA) and Agilent Technologies (CA, USA); and PTFE filters (15-mm diameter, 0.2-μm pore size) were purchased from Teknokroma (Barcelona, Spain).
LC–MS/MS Analysis
The LC–MS/MS system (Fig. 1) consisted of an ACQUITY UPLC and a XEVO TQ-S tandem quadrupole mass spectrometer (Waters, Milford, MA, USA) with an X SELECT C18 column (2.1 mm × 150 mm, 3.5 μm, Waters, Dublin, Ireland) operated at 40 °C. The mobile phases employed were formic acid/acetonitrile/H2O (0.1 : 5 : 95%, v/v/v) (A) and formic acid/H2O/acetonitrile (0.1 : 5 : 95%, v/v/v) (B). The flow rate was 0.3 mL·min−1 and the gradient elution program was as follows: 15% B (0–3 min), 15–95% B (3–8 min), 95% B (8–10 min), 95–15% B (10–10.1 min), and 15% B (10.1–12 min). The total chromatography run time was 12 min. The mass spectrometer was operated under positive electrospray ionization (ESI+) mode for all analytes, with the same capillary voltage (3.5 kV), desolvation and source temperature (350 °C), and cone and desolvation gas flow (60 L·h−1 and 600 L·h−1, respectively). Data collection was implemented in multiple reaction monitoring (MRM) mode using MassLynx software (Waters, UK).


LC–MS/MS chromatograms of target compounds in flatfish (50 μg kg−1 for sulfonamides, ormethoprim, and trimethoprim; 2 μg kg−1 for dapsone and monoacetyldapsone)
Sample Preparation
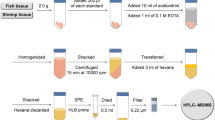
We randomly collected 11 kinds of species (n = 54), viz., abalone (n = 5), catfish (n = 5), eel (n = 7), flatfish (n = 6), flounder (n = 3), loach (n = 7), rockfish (n = 6), salmon (n = 7), and shrimp (n = 8) from fish markets purchased between April and July 2020 in Cheongju, Republic of Korea. The edible tissues (over 500 g) of fish and shrimp were homogenized and stored in a freezer (−20 °C) until residue analysis. A portion of the homogenized samples (2 g) were weighed and transferred to 50-mL polypropylene centrifuge tubes. For each sample, 10 mL of acetonitrile was added. Next, 2 g of magnesium sulfate and 1 g of sodium acetate were added to the samples and shaken for 10 min. Each tube was then centrifuged at 4700×g for 10 min at 4 °C. The supernatant (10 mL) was transferred to a 50-mL polypropylene tube containing 300 mg of C18 as d-SPE sorbent. The mixture was shaken for 1 min and then centrifuged at 4700×g for 10 min at 4 °C. Thereafter, a 5-mL aliquot of the supernatant was pipetted and transferred to another tube, and the solvent was completely evaporated under a stream of nitrogen at 40 °C. The residues were then redissolved in 1.0 mL of 50% methanol and filtered through a 0.2-μm PTFE filter. Subsequently, an aliquot (5 μL) was injected into the LC–MS/MS system (Fig. 2).

Sample preparation procedure for target compounds
Method Validation
Method validation was conducted following the Codex guidelines (CAC/GL-71) to include selectivity, accuracy, precision, linearity, matrix effects, LOD, and LOQ (CCRVDF 2012). In this study, the target testing levels of antibiotics with reported MRLs were validated for each compound in each matrix (flatfish, eel, and shrimp) at three different concentrations (25, 50, and 100 μg kg−1), and unauthorized compounds (i.e., dapsone and monoacetyldapsone) were validated at three different concentrations (1, 2, and 10 μg kg−1) using five replicates for each concentration per day. Flatfish, eel, and shrimp were used as representative matrices [flatfish (lean fish), eel (fatty fish), and shrimp (crustaceans)] to develop a method widely applicable to fish and shrimp matrices (Gibbs et al. 2018). The abalone, flounder, rockfish, and salmon samples were determined based on the flatfish calibration curve; catfish and loach samples were identified using the eel calibration curve; and shrimp samples were processed using the shrimp calibration curve. The intra-day precision test was performed using five replicate measurements for the three concentrations. For assessment of the inter-day precision (reproducibility), the three target concentrations were evaluated for each matrix (n = 5) over three consecutive days. Linearity was evaluated using ordinary least squares expressed by the coefficient (r2) at six different concentration levels in a matrix-matched calibration curve (n = 5). The matrix effect was evaluated by ionic suppression and enhancement, comparing the calibration curves for all analytes prepared in the 50% methanol solvent mixture and in each matrix, separately. Finally, the LOD and LOQ were determined as the lowest concentrations to detect and quantify the analytes, respectively. By injecting the lowest concentrations of standard in each sample, the signal-to-noise (S/N) ratios ≥3 and ≥ 10 were defined as the LOD and LOQ, respectively.
Decision limit (CCα) and decision capability (CCβ) were determined using the ISO 11843 approach, as recommended by the Commission Decision 2002/657/ EC (EC 2002). The CCα was calculated for flat fish, eel, and shrimp using the mean result of 20 different samples spiked at the MRL and adding 1.64 times the standard deviation of the mean. Dapsone was the only exception because it was treated as a banned compound and therefore 2.33 times the within-laboratory reproducibility was added. The CCβ was calculated by adding 1.64 times the within-laboratory reproducibility to the CCα.
Matrix blanks, spiked blanks at MRL levels, and matrix-matched standards at the same concentration were analyzed for each batch of samples to check the reliability of the proposed method. Inter-laboratory validation from four laboratories was conducted to evaluate the ruggedness of the method. Each sample was prepared and analyzed following the same analytical procedure at each of the four participating laboratories using individual LC–MS/MS systems. The accuracy and precision of the five replicate measurements per laboratory are expressed as recovery (%) and coefficient of variation (CV; %).
Results and Discussion
Optimization of LC–MS/MS Analysis
Identification of the precursor and product ions and optimization of the instrument parameter settings were performed using individual standard solutions. The most sensitive transitions used for quantitation and confirmation in MRM mode are listed in Table 1. All compounds exhibited better responses in ESI+ mode. Following the appropriate selection of precursor ions, product ion scans were then conducted to verify the collision energies, which were selected to obtain the maximum intensity of the obtained fragment ions. Four solvent systems were then assessed for reconstitution of the final extract prior to LC–MS/MS injection: (1) 50% methanol, (2) 50% acetonitrile, (3) 1 mM ammonium formate in 50% methanol, and (4) 1 mM ammonium formate in 50% acetonitrile. Among these four solvent mixtures, 50% methanol/50% water was the most effective solvent mixture for reconstitution.
To obtain optimal separation conditions, two C18 columns (X Bridge and Capcell Core) were assessed according to separation efficiency, retention time, and baseline shape. The X Bridge column provided a better peak shape with higher intensity than the Capcell Core column. Since the effect of the mobile phase composition on the ionization efficiency for LC–MS/MS analysis is significant (Cho et al. 2013), four mobile phase combinations were tested: (1) 0.1% formic acid in acetonitrile/0.1% formic acid in H2O, (2) 0.1% formic acid in methanol/0.1% formic acid in H2O, (3) formic acid/H2O/acetonitrile (0.1 : 5 : 95%, v/v/v)/formic acid/acetonitrile/H2O (0.1 : 5 : 95%, v/v/v), and (4) formic acid/H2O/methanol (0.1 : 5 : 95%, v/v/v)/formic acid/methanol/H2O (0.1 : 5 : 95%, v/v/v). Ultimately, mobile phase composition (3) was most promising, prompting our use of formic acid/acetonitrile/H2O (0.1 : 5 : 95%, v/v/v) (A) and formic acid/H2O/acetonitrile (0.1 : 5 : 95%, v/v/v) (B), as these compositions provided well-resolved peaks with high intensity. Figure 1 shows the representative ion chromatograms of the target compounds that were analyzed by injecting each standard solution in flatfish samples at the corresponding 2 µg kg−1 (dapsone) or 50 μg kg−1.
Sample Preparation Methods
Sample preparation involved an organic solvent extraction step to extract analytes from fish samples, followed by a cleanup step to remove interfering substances from the solvent extract (Nunes et al. 2018; Zhang et al. 2011). Acetonitrile is typically used as an extraction solvent for analysis of contaminants and residues in food samples because no matrix interference occurs. In addition, acetonitrile can be easily separated from water by salting out. In this study, acetonitrile was used to extract the target compounds and precipitate protein in sample matrices (Cai et al. 2008). One of the common strategies to increase the solubility of multi-residue molecules in the organic phase is to optimize the average recoveries by introducing a salting out agent (e.g., sodium acetate, magnesium sulfate, or sodium sulfate) (Nunes et al. 2018; Hou et al. 2014; Kim et al. 2016). Thus, we extracted the sample with 10 mL acetonitrile, 2 g magnesium sulfate, and 1 g sodium acetate. This combination accelerated the separation of target analytes into the acetonitrile solvent. For sample cleanup, we compared the efficiency of sorbents during the cleanup step (primary–secondary amine (PSA) and/or C18 were used), with higher recovery obtained from using solely C18; C18 is widely used for removing fats, sterols, and non-polar interfering substances (Hou et al. 2014; Kim et al. 2020). Consequently, the optimal sample preparation procedure was as follows: (1) 10 mL acetonitrile, 2 g magnesium sulfate, and 1 g sodium acetate for extraction; (2) 300 mg C18 as a cleanup sorbent; (3) 50% methanol for reconstitution; and (4) PTFE syringe filter to protect the analytical instrument prior to injection.
Method Validation Performances
The proposed method was validated according to Codex guidelines (CAC/GL-71-2009) to evaluate its analytical performance by analyzing three types of blank samples and comparing their results to the chromatograms obtained from samples spiked with the sulfonamides, dapsone, ormethoprim, and trimethoprim. The farmed aquatic animal matrices (i.e., flatfish, eel, and shrimp) were previously confirmed to be free of the target compounds; no interfering substances with the same retention time were present in the blank samples.
Figure 1 shows the selectivity of the target analytes in different fish and shrimp matrices. Good linearity (r2 > 0.98) was obtained for all target compounds for the matrix-matched standards at 0, 5, 10, 25, 50, 100, and 200 μg kg−1 (dapsone and monoacetyldapsone at 0, 0.25, 0.5, 1, 2, 10, and 20 μg kg−1), with LOD and LOQ of 0.003–0.8 μg kg−1 and 0.01–2.8 μg kg−1, respectively (Table 2). The LOD and LOQ values in this study are similar to or lower than those from previous studies (Li et al. 2016; Dasenaki and Thomaidis 2015; Lopes et al. 2012). LOQs obtained with the developed method are below the MRL and minimum required performance levels (MRPLs) compared with regulatory agencies guidelines (CAC 2017; EC European Commission 2009b; Ministry of Health, Labour and Welfare, Japan 2014; Park et al. 2020). Therefore, the developed method is suitable for the detection of these compounds in fish and shrimp. Accuracy expressed as recovery (76.1–115%) and precision expressed as CV (0.6–11.96%) are listed in Table 2. Inter-laboratory validation at three other laboratories resulted in average recoveries for all compounds of 74.7–83.2%, 74.5–82.5%, and 72.9–84.8%. The recovery values in the present study are similar to previous studies [i.e., 92–111% (Freitas et al. 2014), 55–113% (Liu et al. 2014), and 74.8–104.7% (Li et al. 2016)]. CV values are in the range of 2.4–10.6% (Freitas et al. 2014), 1.8–8.2% (Liu et al. 2014), and 2.5–8% (Li et al. 2016). Compared with previous studies, the validation results for all analytes were satisfactory to determine the residue levels in farmed aquatic animals.
The CCα and CCβ for each analyte in the different matrices are listed in Table 3. The blank material fortified at the maximum residue limit (for analytes with MRL) or at the lowest possible level (for analytes without MRL) in equidistant steps was used. Because the residues of more than one sulfonamide may be present in a single sample, the MRL value is compared to the summed concentration of all detected sulfonamides. For sulfonamides, CCα is 94.2–114 μg kg−1 and CCβ is 102–129 μg kg−1. In the case of dapsone, ormethoprim, and trimethoprim, the CCα values are 1.3–1.5, 105–111, and 53.3–54 μg kg−1 and the CCβ values are 1.4–1.6, 109–120, and 55.6–58.2 μg kg−1, respectively. The obtained values are satisfactory and demonstrate acceptable analytical performance for residue control of fish and shrimp samples.
Evaluation of Matrix Effect
Matrix-matched calibration curves were prepared from blank samples spiked with a standard solution to determine the linearity. To evaluate the effect of the matrix, the slopes obtained in the matrix-matched calibration curves were compared with those obtained using solvent standards. Matrix effects (ME, %) were calculated as follows:
The signal is enhanced if the value is positive, whereas it is suppressed if the value is negative. The matrix effect can be divided into three categories: (1) soft matrix effect (−20% < ME <20%), (2) medium matrix effect (−50% < ME < −20% and 50% > ME >20%), and (3) strong matrix effect (−50% < ME <50%) (SANTE 2015).
In the three representative matrices, weak and moderate matrix effects (−8.3–68.3%) were observed for most of the tested antibiotics. The calculated matrix effects for all compounds are displayed in Table 4. Signal suppression is low in the proposed method; only two target analytes exhibited signal suppression in the fishery products. Of the three matrices, flat fish had the lowest ME, with 60% of the target compounds producing a soft matrix effect. Almost all of the antibiotics exhibited medium matrix effects, and only two compounds had strong matrix effects in the flat fish matrix. Conversely, shrimp gave the highest signal enhancement/suppression: ten analytes had a medium matrix effect and four compounds resulted in strong matrix effects. For eel, most of the compounds provided medium matrix effects, whereas only three compounds exhibited strong matrix effects. Compared with previous studies, our results have shown that the lower matrix effects of sulfonamides in fish samples than that range from −82 to 81.1% (Shin et al. 2018) and from −87 to 56% (Dasenaki and Thomaidis 2015). Similar results in other food product matrices (e.g., livestock and honey) have also been observed for the compounds investigated in the previous studies (Economou et al. 2012; Dasenaki and Thomaidis 2015; Zhang et al. 2016). These results indicate that sulfonamides in farmed aquatic animal samples possess matrix interferences, and therefore matrix-matched standard curves should be employed for residue analysis. Considering our results of matrix effect, an internal standard is not necessary, as the matrix matched calibration with samples can be used to efficiently compensate for differences in recovery across multiple matrices (Hewavitharana 2011).
Application and Monitoring of Real Samples
To evaluate the applicability of the proposed LC–MS/MS method, the presence of sulfonamides, dapsone, ormethoprim, and trimethoprim residues were monitored in 54 samples collected from a local fish market (Table 5). A total detection rate of 13% (n = 7) was obtained for the sulfonamides, dapsone, ormethoprim, and trimethoprim in the fish and shrimp samples, with five out of 24 compounds detected. Specifically, the detected compounds were sulfadiazine (2 μg kg−1 in one sample), sulfamonomethoxine (23–65 μg kg−1 in two samples), sulfamethoxypyridazine (24–61 μg kg−1 in two samples), sulfasomidin (2 μg kg−1 in one sample), and trimethoprim (1 μg kg−1 in three samples). However, none of the compounds exceeded the MRLs established by the Ministry of Food and Drug Safety in Korea. The present investigation confirmed that the proposed method provides optimal analytical performance for real samples.
Conclusions
In this work, we developed and validated an analytical method for the simultaneous quantification of 24 antibiotics in flatfish, eel, and shrimp using d-SPE with LC–MS/MS. The results of validation indicated that the method complies with Codex and EC guidelines. In addition, the method achieved low limit of quantification (LOQ) for all compounds in three different matrices (flatfish, eel, and shrimp). Furthermore, the developed method is rapid and feasible for detecting 21 sulfonamides and three additional antibiotics (dapsone, ormethoprim, and trimethoprim). In real sample analysis, five out of the 24 antibiotics were detected from seven out of 54 samples were detected. The residue concentrations did not exceed the Korean MRL. The proposed method can be used to successfully perform routine analysis of antibiotic residues in farmed aquatic animal products, thus significantly contributing to the development of multi-residue analysis.
References
Baran W, Adamek E, Ziemiańska J, Sobczak A (2011) Effects of the presence of sulfonamides in the environment and their influence on human health. J Hazard Mater 196:1–15. https://doi.org/10.1016/j.jhazmat.2011.08.082
CAC Codex Alimentarius Commission (2015) 38th session of the Codex veterinary drug residue in food online database. Retrieved from http://www.fao.org/fao-who-codexalimentarius/standards/veterinary-drugs-mrls/en/
CAC Codex Alimentarius Commission (2017) Maximum residue limits (MRLs) and risk management recommendations (RMRs) for residues of veterinary drugs in foods. CAC, Rome, Italy, CAC/MRL:2–2017
Cai Z, Zhang Y, Pan H, Tie X, Ren Y (2008) Simultaneous determination of 24 sulfonamide residues in meat by ultra-performance liquid chromatography tandem mass spectrometry. J Chromatogr A 1200:144–155. https://doi.org/10.1016/j.chroma.2008.05.095
CCRVDF Codex Committee on Residues of Veterinary Drugs in Foods (2012) Guidelines for the design and implementation of national regulatory food safety assurance programme associated with the use of veterinary drugs in food producing animals (CAC/GL 71–2009). Codex Alimentarius Commission (CAC), Rome, pp 1–30
Cho SK, Abd El-Aty AM, Park KH, Park J-H, Assayed ME, Jeong Y-M, Park Y-S, Shim J-H (2013) Simple multiresidue extraction method for the determination of fungicides and plant growth regulator in bean sprouts using low temperature partitioning and tandem mass spectrometry. Food Chem 136:1414–1420. https://doi.org/10.1016/j.foodchem.2012.09.068
Dasenaki ME, Thomaidis NS (2015) Multi-residue determination of 115 veterinary drugs and pharmaceutical residues in milk powder, butter, fish tissue and eggs using liquid chromatography–tandem mass spectrometry. Anal Chim Acta 880:103–121. https://doi.org/10.1016/j.aca.2015.04.013
EC European Commission (2002) Council directive 2002/657/EC of 12 August 2002 implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off J Eur Communities L221:8–36
EC European Commission (2009a) On pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin (Reg. EC no. 37/2010). Off J Eur Comm L15:1–72
EC European Commission (2009b) Council regulation (EEC) no. 470/2009 of 6 May 2009 laying down community procedures for the establishment of residue limits of pharmacologically active substances in foodstuffs of animal origin, repealing council regulation (EEC) no. 2377/90. Off J Eur Comm L152:11–22
Economou A, Petraki O, Tsipi D, Botitsi E (2012) Determination of a liquid chromatography–tandem mass spectrometry method for the determination of sulfonamides, trimethoprim and dapsone in honey and validation according to commission decision 2002/657/EC for banned compounds. Talanta 97:32–41. https://doi.org/10.1016/j.talanta.2012.03.058
Freitas A, Leston S, Rosa J, Castilho MC, Barbosa J, Rema P, Pardal MA, Ramos F (2014) Multi-residue and multi-class determination of antibiotics in gilthead sea bream (Sparus aurata) by ultra high-performance liquid chromatography-tandem mass spectrometry. Food Addit Contam A 31:817–826. https://doi.org/10.1080/19440049.2014.891764
Gibbs RS, Murray SL, Watson LV, Nielsen BP, Potter RA, Murphy CJ (2018) Development and validation of a hybrid screening and quantitative method for the analysis of eight classes of therapeutants in aquaculture products by liquid chromatography–tandem mass spectrometry. J Agric Food Chem 66:4997–5008. https://doi.org/10.1021/acs.jafc.7b05357
Hewavitharana AK (2011) Matrix matching in liquid chromatography–mass spectrometry with stable isotope labelled internal standards—is it necessary? J Chromatogr A 1218:359–361. https://doi.org/10.1016/j.chroma.2010.11.047
Hoff RB, Pizzolato TM, Peralba MCR, Díaz-Cruz MS, Barceló D (2015) Determination of sulfonamide antibiotics and metabolites in liver, muscle and kidney samples by pressurized liquid extraction or ultrasound-assisted extraction followed by liquid chromatography–quadrupole linear ion trap-tandem mass spectrometry (HPLC–QqLIT-MS/MS). Talanta 134:768–778. https://doi.org/10.1016/j.talanta.2014.10.045
Hou X, Lei S, Qiu S, Guo L, Yi S, Liu W (2014) A multi-residue method for the determination of pesticides in tea using multi-walled carbon nanotubes as a dispersive solid phase extraction absorbent. Food Chem 153:121–129. https://doi.org/10.1016/j.foodchem.2013.12.031
Jia W, Shi L, Chu X (2018) Untargeted screening of sulfonamides and their metabolites in salmon using liquid chromatography coupled to quadrupole Orbitrap mass spectrometry. Food Chem 239:427–433. https://doi.org/10.1016/j.foodchem.2017.06.143
Kang H-S, Lee S-B, Shin D, Jeong J, Hong J-H, Rhee G-S (2018) Occurrence of veterinary drug residues in farmed fishery products in South Korea. Food Control 85:57–65. https://doi.org/10.1016/j.foodcont.2017.09.019
Kang H-S, Han S, Cho B-H, Lee H (2019a) Risk-based approach to develop a national residue program: prioritizing the residue control of veterinary drugs in fishery products. Fish Aquat Sci 22:29. https://doi.org/10.1186/s41240-019-0143-2
Kang H-S, Kwon NJ, Jeong J, Lee K, Lee H (2019b) Web-based Korean maximum residue limit evaluation tools: an applied example of maximum residue limit evaluation for trichlorfon in fishery products. Environ Sci Pollut Res 26:7284–7299. https://doi.org/10.1007/s11356-019-04314-y
Kim K, Park D, Kang G, Kim T, Yang Y, Moon S, Choi E, Ha D, Kim E, Cho B (2016) Simultaneous determination of plant growth regulator and pesticides in bean sprouts by liquid chromatography–tandem mass spectrometry. Food Chem 208:239–244. https://doi.org/10.1016/j.foodchem.2016.04.002
Kim J, Park H, Kang H-S, Cho B-H, Oh J-H (2020) Comparison of sample preparation and determination of 60 veterinary drug residues in flatfish using liquid chromatography-tandem mass spectrometry. Molecules 25:1206. https://doi.org/10.3390/molecules25051206
Li J, Liu H, Zhang J, Liu Y, Wu L (2016) A novelty strategy for the fast analysis of sulfonamide antibiotics in fish tissue using magnetic separation with high-performance liquid chromatography–tandem mass spectrometry. Biomed Chromatogr 30:1331–1337. https://doi.org/10.1002/bmc.3693
Liu S, Du J, Chen J, Zhao H (2014) Determination of 19 antibiotic and 2 sulfonamide metabolite residues in wild fish muscle in mariculture areas of Laizhou Bay using accelerated solvent extraction and high performance liquid chromatography-tandem mass spectrometry. Chin J Chromatogr 32:1320–1325. https://doi.org/10.3724/sp.j.1123.2014.08032
Lopes RP, Reyes RC, Romero-González R, Martínez Vidal JL, Frenich AG (2012) Multiresidue determination of veterinary drugs in aquaculture fish samples by ultra high performance liquid chromatography coupled to tandem mass spectrometry. J Chromatogr B 895–896:39–47. https://doi.org/10.1016/j.jchromb.2012.03.011
McDonald M, Mannion C, Rafter P (2009) A confirmatory method for the simultaneous extraction, separation, identification and quantification of tetracycline, sulphonamide, trimethoprim and dapsone residues in muscle by ultra-high-performance liquid chromatography–tandem mass spectrometry according to commission decision 2002/657/EC. J Chromatogr A 1216:8110–8116. https://doi.org/10.1016/j.chroma.2009.05.092
Ministry of Health, Labour and Welfare, Japan (2014) The Japanese positive list system for agricultural chemical residues in foods, Tokyo, Japan. http://www.ffcr.or.jp/ zaidan/FFCRHOME.nsf/pages/MRLs-p. Accessed on March 10, 2015
Nicosia A, Celi M, Vazzana M, Damiano MA, Parrinello N, D’Agostino F, Avellone G, Indelicato S, Mazzola S, Cuttitta A (2014) Profiling the physiological and molecular response to sulfonamidic drug in Procambarus clarkii. Comp Biochem Physiol C Toxicol Pharmacol 166:14–23. https://doi.org/10.1016/j.cbpc.2014.06.006
Nunes KSD, Assalin MR, Vallim JH, Jonsson CM, Queiroz SCN, Reyes FGR (2018) Multiresidue method for quantification of sulfonamides and trimethoprim in tilapia fillet by liquid chromatography coupled to quadrupole time-of-flight mass spectrometry using QuEChERS for sample preparation. J Anal Methods Chem 2018:4506754–4506710. https://doi.org/10.1155/2018/4506754
Park HJ, Kim JH, Kang H-S, Cho B-H, Oh JH (2020) Multi-residue analysis of 18 dye residues in animal products by liquid chromatography-tandem mass spectrometry. J Food Hyg Saf 35:109–117. https://doi.org/10.13103/JFHS.2020.35.2.109
Reeves PT (2012) Antibiotics: groups and properties. Chemical analysis of antibiotic residues in food. Wiley, New Jersey, pp 30–31
SANTE (2015) Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed European Commission document no SANTE/11945/2015
Serrano PH (2005) Responsible use of antibiotics in aquaculture. FAO, Rome 469
Shin D, Kang H-S, Jeong J, Kim J, Choe WJ, Lee KS, Rhee G-S (2018) Multi-residue determination of veterinary drugs in fishery products using liquid chromatography-tandem mass spectrometry. Food Anal Methods 11:1815–1831. https://doi.org/10.1007/s12161-018-1179-0
Song C, Li L, Zhang C, Kamira B, Qiu L, Fan L, Wu W, Meng S, Hu G, Chen J (2017) Occurrence and human dietary assessment of sulfonamide antibiotics in cultured fish around Tai Lake, China. Environ Sci Pollut Res 24:17493–17499. https://doi.org/10.1007/s11356-017-9442-2
USDA United States Department of Agriculture (2017) Maximum residue limits (MRL) database. Retrieved from https://www.fas.usda.gov/maximum-residue-limits-mrl-database
Zhang K, Wong JW, Yang P, Tech K, DiBenedetto AL, Lee NS, Hayward DG, Makovi CM, Krynitsky AJ, Banerjee K, Jao L, Dasgupta S, Smoker MS, Simonds R, Schreiber A (2011) Multiresidue pesticide analysis of agricultural commodities using acetonitrile salt-out extraction, dispersive solid-phase sample clean-up, and high-performance liquid chromatography–tandem mass spectrometry. J Agric Food Chem 59:7636–7646. https://doi.org/10.1021/jf2010723
Zhang Z, Li X, Ding S, Jiang H, Shen J, Xia X (2016) Multiresidue analysis of sulfonamides, quinolones, and tetracyclines in animal tissues by ultra-high performance liquid chromatography-tandem mass spectrometry. Food Chem 204:252–262. https://doi.org/10.1016/j.foodchem.2016.02.142
Zhu YI, Stiller MJ (2001) Dapsone and sulfones in dermatology: overview and update. J Am Acad Dermatol 45:420–434. https://doi.org/10.1067/mjd.2001.114733
Funding
This study was supported by grants (No. 19161MFDS581 and 20161MFDS623) from the Ministry of Food and Drug Safety in South Korea in 2019 and 2020.
Author information
Authors and Affiliations
Contributions
SYC: performed experimental work and drafted of manuscript
H-SK: study designed and wrote and revised manuscript
Corresponding author
Ethics declarations
Ethical Approval
This article does not contain any studies with human participants or animals by any of the authors.
Informed Consent
Not applicable.
Conflict of Interest
SYC and H-SK declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Choi, S.Y., Kang, HS. Multi-Residue Determination of Sulfonamides, Dapsone, Ormethoprim, and Trimethoprim in Fish and Shrimp Using Dispersive Solid Phase Extraction with LC–MS/MS. Food Anal. Methods 14, 1256–1268 (2021). https://doi.org/10.1007/s12161-021-01965-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-021-01965-x