
Abstract
In the present study, an elevated temperature homogeneous liquid extraction coupled with ionic liquid‑based dispersive liquid–liquid microextraction was developed to extract five carbamate pesticides (pirimicarb, carbaryl, methiocarb, aldicarb, and carbofuran) from packed fruit juice samples. The method consisted of a two-step procedure in which cyclohexylamine was used in order to extract the target analytes. In the first step, the analytes were extracted from a sample solution thermostated at elevated temperature (80 °C) into a water-immiscible organic solvent (cyclohexylamine). In the second step, the separated organic phase was mixed with 1-butyl-3-methylimidazolium chloride (as the solvent for extraction) and the mixture was spread into deionized water. In this step, cyclohexylamine controlled the solubility of 1-butyl-3-methylimidazolium chloride in the aqueous phase and acted as a dispersive solvent and a phase separation agent, simultaneously. After centrifugation, we removed all the gathered organic phases and analyzed them by using high-performance liquid chromatography diode-array detector. Figure of merits of the method including the limits of detection and quantification, enrichment factors, extraction recoveries, and relative standard deviations were obtained within ranges 0.4–3.9 and 1.5–13.2 ng mL−1, 147–217, 59–87%, and 3.3–5.5%, respectively, indicating that the method was successful in determining the analytes.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since the production and protection of manufactured products are carried out in parallel, there is always a need to preserve the agricultural productions (Wasim Aktar et al. 2009). The use of pesticides is inevitable in order to protect agricultural products against pests. During the last decades, pesticides have become an essential part of agricultural activities throughout the world. Pesticides are classified into different categories with several applications (Chandra Yadav and Linthoingambi Devi 2017). Classification of pesticides is mainly performed by considering their structure and functional groups. As a result, some pesticides including triazole, triazine, organophosphorus, acidic, and carbamate are considered. For this purpose, some pesticides like carbamates have broad applications in houses, gardens, and farms against insects based on their fairly short lifespan, impact, and wide range of biological activity (Yongnian et al. 2005; Solé et al. 2003). However, the toxicity of carbamates causes some concerns about their usage as a potential hazard for consumers (Bedendo et al. 2012; Msagati and Mamba 2012). Therefore, many organizations have established strict instructions for their use in agriculture and have defined the highest residue limits (MRLs) in agricultural crops in the ranges of 0.01–0.05 mg kg−1 (http://ec.europa.eu/food/plant/pesticides/eu-pesticides-database n.d.). Since the MRLs are generally very low, sensitive determination procedures are needed for pesticide analyte residues in food samples. Chromatographic methods such as high-performance liquid chromatography (HPLC) equipped with diode-array detector (DAD) (Salvatierra-stamp et al. 2018; Zhou and Fang 2015), mass spectrometry (MS) (Fernández et al. 2000; Leça et al. 2014), mass spectrometry/mass spectrometry (MS/MS) (Moreno-González et al. 2013), gas chromatography (GC) coupled with MS (Zhang and Kee Lee 2006; Mohebbi et al. 2018), MS/MS (Lachenmeier et al. 2006; Chen et al. 2010), and micellar electrokinetic chromatography (Santalad et al. 2010), as well as other obtainable methods such as enzyme-linked immunosorbent assays (Wang et al. 2005) and ion mobility (Ghoraba et al. 2018), have been used for this purpose. Although these methods are sufficiently sensitive for the determination of carbamates, the complexity of real samples prevents their direct use in analyzing the sample analytes. Accordingly, sample preparation procedures have attracted more attention before real sample analysis (Mitra 2003). Sample preparation procedures are usually based on solvent extraction methods (SEM) and among the SEMs, microextraction procedures are used in different samples. Microextraction techniques are classified into two major groups including liquid-phase microextraction (LPME) (Afshar Mogaddam et al. 2020) and solid-phase microextraction (SPME) (Llompart et al. 2019). LPME is based on three main modes including hollow fiber liquid-phase microextraction (HF-LPME) (Salvatierra-stamp et al. 2018), single-drop microextraction (SDME) (Tang et al. 2018), and dispersive liquid–liquid microextraction (DLLME) (Rezaee et al. 2006). DLLME was innovated in 2006 by Assadi and coworkers and developed rapidly for the determination of many compounds (Farajzadeh et al. 2018; Wang et al. 2019). DLLME extracts the analytes using a three-phase system consist of a water-miscible organic solvent (dispersive solvent), water-immiscible organic solvent (extraction solvent), and the sample solution. In this step, the extraction solvent is dispersed in all parts of the solution with the aid of a dispersive solvent, and the analytes are extracted in the form of tiny extractant droplets. After centrifugation, the collected extraction solvent is used in the following determination system. DLLME has some advantages like operation simpleness, low cost, and high efficiency and concentration factor. However, the use of chlorinated organic solvents as the extraction solvent is the main drawback of DLLME procedures. Therefore, in recent years, more attention has been directed to the use of ionic liquids (ILs) as a replacement for organic extraction solvents (Unsal et al. 2015; Unsal et al. 2019; Yelmaz and Soylak 2013; Farajzadeh et al. 2016). However, there are many ILs with high solubility in water which are not used in DLLME. These ILs are cheaper and more accessible than water-immiscible ones. Yet, the presence of some other compounds may alter the solubility of ILs in water and make them usable in DLLME method.
In this research, a liquid–liquid extraction (LLE) coupled to DLLME procedure was followed to extract some carbamate pesticides from the packed samples of fruit juice followed by HPLC-DAD. For this purpose, an organic solvent, e.g., cyclohexylamine (water-immiscible at high temperatures and water-miscible at room temperature), was used as an extraction solvent and disperser solvent in LLE and DLLME procedures, respectively. The LLE extractant was blended with a water-miscible IL and the mixture was used in DLLME procedure. The main feature of this procedure is the role of cyclohexylamine as an extraction solvent in LLE and a dispersive solvent as well as a phase separation agent in DLLME. All of the efficient factors were examined and optimized.
Experimental
Chemicals and Reagents
The studied carbamates consisting of pirimicarb, carbaryl, methiocarb, carbofuran, and aldicarb with purity above 98.5% were bought from Dr. Ehrenstorfer Company (Augsburg, Germany). The tested ILs 1-butyl-3-methylimidazolium bromide ([BMIM]Br), 1-ethyl-3-methylimidazolium chloride ([EMIM]Cl), and 1-butyl-3-methylimidazolium chloride ([BMIM]Cl) were bought from Sigma-Aldrich (St. Louis, MO, USA). Cyclohexylamine, NaCl, sodium hydroxide, and hydrochloric acid were obtained from Merck (Darmstadt, Germany). Deionized water was prepared using a Milli–Q water system (Millipore, Billerica, MA, USA). HPLC grade water and acetonitrile were obtained from Chemlab (Zedelgem, Belgium). The studied pesticides were dissolved in methanol to prepare a stock solution at a concentration of 50 mg L−1of (each analyte), and the working solutions were prepared from this solution by proper dilutions with HPLC-grade water.
Samples
Commercially packed grape, apple, sour cherry, pomegranate, and apricot juices were bought from local vendors (Tabriz, East Azarbaijan, Iran). All of the samples were diluted with deionized water at a ratio of 1:4 and then utilized in the developed method.
Instrumentation
Liquid chromatographic analysis was conducted on an Agilent 1200 (Agilent Technologies, Santa Clara, CA, USA) equipped with a degasser, quaternary pump, and DAD. Separation was performed on a DC18-3UM analytical column (150 mm × 4.6 mm, 5 μm particle size) (Waters, Milford, MA, USA), and the analytes were eluted with a mixture of methanol: water (65:35, v/v) at a flow rate of 0.7 mL min−1. Detection of the pesticides was done at 205 nm for carbofuran and carbaryl, at 223 nm for pirimicarb, and at 228 nm for aldicarb and methiocarb.
Pretreatment Procedure
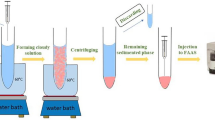
In total, 5.0-mL deionized water spiked at 25 ng mL−1 (each pesticide) or a diluted sample solution was transferred into a 10-mL glass test tube, and 0.375 g NaCl (7.5%, w/v) was dissolved in it. The tube was placed in a water bath (adjusted at 80 °C) for 3.0 min. Then 0.75 mL cyclohexylamine was added and vortexed for 3 min. The solution was centrifuged for 7 min at 4000 rpm, and 1.0 mL of collected cyclohexylamine was mixed with [BMIM]Cl (55 μL). The mixture was rapidly injected into 5-mL deionized water. The cloudy solution was centrifuged for 5 min at 4000 rpm. All the sedimented phase (20 ± 1 μL) was removed and injected into the separation system.
Calculation of Enrichment Factor and Extraction Recovery
Enrichment factor (EF) is considered the analyte concentration ratio in the final phase (Cfin) to the initial concentration (C0) in the sample.
Extraction recovery (ER) refers to the percentage of the total amount of the analyte (n0) which is transferred into the extractive phase (nfin).
where Vfin and Vaq are the volumes of the final organic phase and aqueous phase, respectively.
Results and Discussion
To obtain the optimized extraction conditions, the “one-variable-at-a-time” technique was applied to evaluate the efficient parameters. In this step, different parameters such as type and volume of the extraction solvent (in two steps), salt effect, temperature, pH, speed of centrifugation, and time were optimized.
Optimization of Parameters in LLE Step
Selection of Extraction Solvent
In this work, the analytes were extracted into a water-miscible (at room temperature) and water-immiscible (at high temperature) solvent by performing an LLE procedure in an elevated temperature. In LLE, the extractive phase should not be miscible with the sample solution in order to obtain a two-phase system; however, in the present work, a water-miscible organic solvent was used as an extraction solvent from the aqueous phase. For this purpose, a two-phase system was formed by changing the temperature. As a result, three important factors namely high efficiency for the analytes, a different density from water, and low solubility in the aqueous phase in an elevated temperature were considered. However, the selected extraction solvent had to be miscible with both the aqueous phase and the extraction solvent used in DLLME step. Taking these parameters into account, cyclohexylamine was examined as a possible extraction solvent. For the evaluation of cyclohexylamine as the extraction solvent in LLE and disperser solvent in DLLME, 5-mL deionized water spiked at the concentration of 25 ng mL−1 (each analyte) was transferred into a glass test tube and placed in a water bath thermostated at 70 °C for 4 min. Then, we added 1.0 mL cyclohexylamine to the solution and vortexed the mixture for 1 min. The obtained results showed that the 1.2-mL organic phase (cyclohexylamine saturated phase) was separated as the upper phase. The gathered phase was used in the following DLLME method. It is remarkable that some portion of the water was dissolved in the organic solvent which contained the analytes. The results showed that the ERs for all analytes were in the range of 37–49%.
Salt Addition
Adding salt is generally used to enhance the distribution of the analytes into an organic phase by decreasing their solubility in the aqueous phase simultaneously. Furthermore, salt addition can also reduce the solubility of the extraction phase in the aqueous solution. The influence of salt addition on the performance of the method was achieved by the addition of different amounts of NaCl to the aqueous phase in the range of 0.0–12.5%, w/v, while the other parameters remained constant. The plot of peak area vs. NaCl concentration (Fig. 1) shows that the peak areas increase up to 7.5%, w/v, NaCl and then decrease by increasing the NaCl concentration. It is noteworthy that the tests were performed using different volumes of cyclohexylamine to obtain 1.2 mL of the supernatant phase as 1.0, 0.95, 0.82, 0.75, 0.68, and 0.55 mL for 0, 5.0, 7.5, 10.0, and 12.5%, w/v, NaCl, respectively.

Optimization of NaCl concentration on the performance of LLE step. Extraction conditions in LLE step: sample, 5-mL deionized water spiked with the analytes at 25 ng mL−1 (each analyte); aqueous phase temperature (time), 70 °C (4 min); extraction solvent (volume) cyclohexylamine (1.2 mL); vortexing time, 1 min; and centrifugation speed (time) 4000 rpm (5 min). Extraction conditions in DLLME step: disperser solvent, 1.0 mL of the supernatant phase obtained from the previous step; extraction solvent type (volume) in DLLME step, [BMIM]Br (55 μL); and volume of aqueous phase, 5-mL deionized water, and centrifugation speed (min), 4000 rpm (5 min). The error bars show the lowest and highest rates of the three determinations
Optimization of Cyclohexylamine Volume
The volume of cyclohexylamine has the main impact on the performance of the method via changing the volume ratio of aqueous to the organic phase. It is obvious that at low volumes of the organic phase, ER% decreases whereas EF increases, simultaneously. Evaluation of the cyclohexylamine volume effect on the performance of the method was carried out by subjecting various volumes of cyclohexylamine in the range of 0.50–2.00 mL (at 0.25-mL intervals) to the same procedures. It is noteworthy that by raising the cyclohexylamine volume, the gathered organic phase volume rose from 1.0 to 2.7 mL. The obtained results (Fig. 2) show that the analytical signals increase up to 0.75 mL and then decrease. Therefore, 0.75 mL of cyclohexylamine was used for further experiments.

Optimization of cyclohexylamine volume. Extraction conditions are the same as those used in Fig. 1, except NaCl concentration which was 7.0% (w/v)
Optimization of Aqueous Phase Temperature
In this work, aqueous phase temperature played a significant role in performing the developed method. Increasing the temperature had two effects in this study. The first one was decreasing cyclohexylamine solubility in the aqueous solution and the feasibility of LLE procedure at high temperature, whereas cyclohexylamine and aqueous phase formed a homogeneous solution at room temperature. The second one was the improvement of mass transfer rates of the analytes. The effect of temperature was studied in the range of 50–90 °C. We noticed that at temperatures below 50 °C, cyclohexylamine was not separated from the solution and the method failed to work. Figure 3 shows the variations in peak areas versus aqueous phase temperature. The results revealed that the analytical signals rise up to 80 °C and then stay nearly constant. At lower temperatures, the diffusion coefficients and mass transfer of the analytes and analytical signals decrease simultaneously. Subsequently, the aqueous phase temperature was adjusted at 80 °C in the next experiments.

Optimization of aqueous phase temperature. Extraction conditions are identical to those used in Fig. 2 except cyclohexylamine volume which was fixed at 0.75 mL
Optimization of Heating Time
The other effective parameter in the efficiency of the method is the heating time. The heating time may have two roles in this method. The heating time affects the mass transfer rate of the analytes from the aqueous solution into the extraction solvent and assists in the separation of cyclohexylamine. To optimize the heating time, different experiments in the range of 0.5–5.0 min (at 0.5-min intervals) were performed while the other conditions remained constant. The results in Fig. 4 indicate that when the heating time rises from 0.5 to 3.0 min, the analytical signals rise, simultaneously. Nevertheless, the extraction efficiency shows no significant improvement when the heating time goes up from 3.0 to 5.0 min. Thus, 3.0 min was selected as the optimal heating time.

Optimization of heating time. Extraction conditions are the same as those applied in Fig. 3, except aqueous phase temperature which was set at 80 °C
Optimization of Parameters in DLLME Method
Selection of Extraction Solvent Type
Selection of a proper extraction solvent is considered a critical parameter in DLLME method. The extraction solvent should have some qualities such as desirable chromatographic behavior, extraction capability of the analytes, and different density from the aqueous phase. In this method, water-miscible ILs were used as possible extraction solvents. The main parameter in their selection was that their solubility decreased in the presence of cyclohexylamine. To study the kind of extraction solvent in the performance of the developed method, [BMIM]Cl, [BMIM]Br, and [EMIM]Cl were subjected to the same procedure. It is noteworthy that the experiments were conducted using various volumes of the studied ILs to obtain the same volume of the sedimented phase (20 ± 1 μL). For this purpose, 55, 72, and 105 μL of [BMIM]Cl, [BMIM]Br, and [EMIM]Cl were used, respectively. Based on the obtained results in Fig. 5, [BMIM]Cl shows the highest analytical signals among the examined ILs. Therefore, it was chosen as the extractant in the next experiments.

Selection of extraction solvent in DLLME step. Extraction conditions are the same as those used in Fig. 4, except 3 min was used as a heating time
Optimization of Extraction Solvent Volume
In the developed method, the extraction solvent volume is an important parameter which can affect the method performance by changing the volume ratio of the aqueous phase to the organic phase. High EFs and low limits of detection (LODs) can be obtained at low volumes of the organic phase. Therefore, the changes in DLLME performance by extraction solvent volume were studied in the range of 55 to 100 μL. The results showed that by increasing the volume of [BMIM]Cl in the mentioned range, the sedimented phase volume rose from 20 to 68 μL, and the peak areas declined. This can be due to the dilution of the analytes in the sedimented phase. Therefore, 55 μL of [BMIM]Cl was selected for the next experiments.
Salt Addition
In extraction procedures, the addition of salt is commonly used for enhancing the efficiency of the method. Dissolving salt in aqueous phase raises the ionic strength of the aqueous phase and reduces the solubility of the extraction solvent and the analytes in the sample solution at the same time. These can strengthen the partitioning of the analytes into the extraction solvent. To assess the salt addition effect, different concentrations of sodium chloride in the range of 0–8.0%, w/v, were separately added to the deionized water, and the method was applied to them. The experiments were performed using different volumes of [BMIM]Cl to achieve the same volume of the sedimented phase (20 ± 1 μL). Therefore, 55, 48, 42, 36, and 30 μL of [BMIM]Cl were used for 0, 2.0, 4.0, 6.0, and 8.0%, w/v, NaCl, respectively. The results indicated that the peak areas decreased from 0 to 8.0%, w/v, NaCl. Therefore, the experiments were done without adding salt.
Study of Other Parameters
The impact of other parameters on the efficiency of the method was studied by altering pH, centrifugation speed, and time in the ranges of 2–12, 2000–5000 rpm, and 5–10 min, respectively. The data indicated that there were no significant differences among the peak areas at pHs, centrifugation speeds, and times in the ranges of 4–10, 3000–5000 rpm, and 7–10 min, respectively. According to these results, there was no need to adjust the solution pH. The centrifugation speed and time were fixed at 4000 rpm and 5 min, respectively.
Quantitative Aspects
Quantitative aspects of the proposed method was examined in terms of LOD (based on signal to noise ratio of 3), limit of quantification (LOQs, based on signal to noise ratio of 10), linear range (LR), determination coefficient (r2), relative standard deviation (RSD), EF, and ER. The data are listed in Table 1. Broad linearities with r2 ≥ 0.9623 were obtained. The LODs and LOQs were within the ranges of 0.4–3.9 and 1.9–13.2 ng mL−1, respectively. Accuracy of the method was studied by analyzing the spiked samples at the concentration of 25 ng mL−1, on the same day (n = 6) and on four different days. The RSDs were found to be lower than 8%. The EFs and ERs for the chosen pesticides ranged from 147 to 217 and 59 to 87%, respectively. These data indicate that analyzing the studied pesticides in aqueous samples can be performed appropriately.
Real Sample Analysis
The applicability of the developed method in determining the selected pesticides was evaluated by analyzing five packed fruit juice samples including apple, grape, pomegranate, sour cherry, and apricot juices under optimum conditions. Figure 6 shows typical HPLC-DAD chromatograms of the standard solution of grape, and pomegranate samples after applying the developed method on them. Comparison of the chromatograms of the standard solution and the samples confirms that all of the samples are free of the analytes. To evaluate matrix effects of the samples and deionized water on the efficiency of the method, the samples were separately spiked with the analytes at three different concentrations (15, 25, and 50 ng mL−1 of each analyte) and the recoveries for the samples in comparison with deionized water are given in Table 2. The data show that the matrices of the samples have no significant impact on the functioning of the presented method. Hence, it is inferred that the proposed method can be applied as an applicable and efficient method for the analysis of the selected analytes in the samples.

Typical HPLC-DAD chromatograms of (I) direct injection of standard solution at concentration of 10 mg L−1, (II) un-spiked grape sample after performing the devised method, (III) un-spiked pomegranate sample after performing the devised method. Peaks identification: 1) pirimicarb, 2) carbaryl, 3) methiocarb, 4) carbofuran, and 5) aldicarb
Comparison of the Method with Other Approaches
The performance of the present method was compared with other previously published sample preparation methods coupled with HPLC-DAD including HF-LPME, SPME, and ultrasound-assisted surfactant-enhanced emulsification microextraction (UASEME) from the viewpoint of LODs, RSDs, EFs, and LRs. As listed in Table 3, the LODs and RSDs of the method are lower than or comparable with those of the other approaches. The method has a broader linear range except for the SPME-HPLC-DAD method. The EFs of the method were higher than or comparable with those of the other methods. Therefore, the method can be applied as an alternative way for the extraction of the selected analytes from fruit juice samples.
Conclusion
In this study, a simple and reliable sample pretreatment method was developed for the extraction and preconcentration of carbamate pesticides from fruit juice samples. The proposed method consisted of a two-step procedure in which LLE was coupled with IL-based DLLME. In the first step, the analytes were extracted into a water-miscible organic solvent (cyclohexylamine) from a sample solution. The sample solution was adjusted at an elevated temperature to form a two-phase system. The extractant used in the LLE step was utilized as a dispersive solvent and phase separation agent in the following DLLME procedure, simultaneously. In the second step, the supernatant phase obtained from the previous step was mixed with a water-miscible IL, and the mixture was used in DLLME. In this method, water-miscible ILs were tested as the possible extraction solvents which are more accessible and cheaper than the other ILs. The method had acceptable precision, low LODs, LOQs, and high ERs and EFs. The method can be applied for the determination of the target analytes in the range of ng mL−1 in fruit juice samples.
Abbreviations
- EF:
-
Enrichment factor
- DLLME:
-
Dispersive liquid–liquid microextraction
- LOQ:
-
Limit of quantification
- LR:
-
Linear range
- LOD:
-
Limit of detection
- IL:
-
Ionic liquid
- LLE:
-
Liquid–liquid extraction
- ER:
-
Extraction recovery
- RSD:
-
Relative standard deviation
References
Afshar Mogaddam MR, Mohebbi A, Pazhohan A, Khodadadeian F, Farajzadeh MA (2020) Headspace mode of liquid phase microextraction: a review. Trends Anal Chem 110:8–14
Bedendo GC, Jardim ICSF, Carasek E (2012) Multiresidue determination of pesticides in industrial and fresh orange juice by hollow fiber microporous membrane liquid–liquid extraction and detection by liquid chromatography–electrospray-tandem mass spectrometry. Talanta 88:573–580
Chandra Yadav I, Linthoingambi Devi N (2017) Environmental science and engineering: pesticides classification and its impact on human and environment, 6th edn. Studium Press LLC, USA
Chen H, Chen R, Li S (2010) Low-density extraction solvent-based solvent terminated dispersive liquid–liquid microextraction combined with gas chromatography-tandem mass spectrometry for the determination of carbamate pesticides in water samples. J Chromatogr A 1217:1244–1248
Farajzadeh MA, Bamorowat M, Afshar Mogaddam MR (2016) Ringer tablet-based ionic liquid phase microextraction: application in extraction and preconcentration of neonicotinoid insecticides from fruit juice and vegetable samples. Talanta 160:211–216
Farajzadeh MA, Shahedi Hojghan A, Afshar Mogaddam MR (2018) Development of a new temperature-controlled liquid phase microextraction using deep eutectic solvent for extraction and preconcentration of diazinon, metalaxyl, bromopropylate, oxadiazon, and fenazaquin pesticides from fruit juice and vegetable samples followed by gas chromatography-flame ionization detection. J Food Compos Anal 66:90–97
Fernández M, Picó Y, Manes J (2000) Determination of carbamate residues in fruits and vegetables by matrix solid-phase dispersion and liquid chromatography–mass spectrometry. J Chromatogr A 871:43–56
Ghoraba Z, Aibaghi B, Soleymanpour A (2018) Ultrasound-assisted dispersive liquid-liquid microextraction followed by ion mobility spectrometry for the simultaneous determination of bendiocarb and azinphos-ethyl in water, soil, food and beverage samples. Ecotoxicol Environ Saf 165:459–466
http://ec.europa.eu/food/plant/pesticides/eu-pesticides-database (n.d.) EU Pesticides database. Part A of Annex I to Reg. 396/2005
Lachenmeier DW, Nerlich U, Kuballa T (2006) Automated determination of ethyl carbamate in stone-fruit spirits using headspace solid-phase microextraction and gas chromatography–tandem mass spectrometry. J Chromatogr A 1108:116–120
Leça JM, Pereira V, Pereira AC, Marques JC (2014) Rapid and sensitive methodology for determination of ethyl carbamate in fortified wines using microextraction by packed sorbent and gas chromatography with mass spectrometric detection. Anal Chim Acta 811:29–35
Llompart M, Celeiro M, García-Jares C, Dagnac T (2019) Environmental applications of solid-phase microextraction. Trends Anal Chem 112:1–12
Mitra S (2003) Sample preparation techniques in analytical chemistry, First edn. Wiley, London
Mohebbi A, Yaripour S, Farajzadeh MA, Afshar Mogaddam MR (2018) Combination of dispersive solid phase extraction and deep eutectic solvent–based air–assisted liquid–liquid microextraction followed by gas chromatography–mass spectrometry as an efficient analytical method for the quantification of some tricyclic antidepressant drugs in biological fluids. J Chromatogr A 1571:84–93
Moreno-González D, Huertas-Pérez JF, García-Campaña AM, Bosque-Sendra JM, Gámiz-Gracia L (2013) Ultrasound-assisted surfactant-enhanced emulsification microextraction for the determination of carbamates in wines by ultra-high performance liquid chromatography–tandem mass spectrometry. J Chromatogr A 1315:1–7
Msagati TA, Mamba BB (2012) Monitoring of N-methyl carbamate pesticide residues in water using hollow fiber supported liquid membrane and solid phase extraction. Phys Chem Earth A/B/C 50:149–156
Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid–liquid microextraction. J Chromatogr A 1116:1–9
Salvatierra-stamp V, Muñiz-Valencia R, Jurado JM, Ceballos-Magaña SG (2018) Hollow fiber liquid phase microextraction combined with liquid chromatography-tandem mass spectrometry for the analysis of emerging contaminants in water samples. Microchem J 140:87–95
Santalad A, Zhou L, Shang F, Fitzpatrick D, Luong JHT (2010) Micellar electrokinetic chromatography with amperometric detection and off-line solid-phase extraction for analysis of carbamate insecticides. J Chromatogr A 1217:5288–5297
Solé S, Merkoçi A, Alegret S (2003) Determination of toxic substances based on enzyme inhibition. Part I. Electrochemical biosensors for the determination of pesticides using batch procedures. Crit Rev Anal Chem 33:89–126
Tang S, Qi T, Dim Ansah P, Nalouzebi Fouemina JC, Kee Lee H (2018) Single-drop microextraction. Trends Anal Chem 108:306–313
Unsal YE, Soylak M, Tuzen M (2015) Ultrasound-assisted ionic liquid-based dispersive liquid-liquid microextraction for preconcentration of patent blue V and its determination in food samples by UV-visible spectrophotometery. Environ Monit Assess 187:203–210
Unsal YE, Soylak M, Tuzen M (2019) Ultrasound-assisted ionic liquid-dispersive liquid-liquid of curcumin in food samples microextraction and its determination. J AOAC Int 102:217–221
Wang S, Yu C, Wang J (2005) Enzyme immunoassay for the determination of carbaryl residues in agricultural products. Food Addit Contam 22:735–742
Wang Q, Chen R, Shatner W, Cao Y, Bai Y (2019) State-of-the-art on the technique of dispersive liquid-liquid microextraction. Ultrason Sonochem 51:369–377
Wasim Aktar M, Sengupta D, Chowdhury A (2009) Impact of pesticides use in agriculture: their benefits and hazards. Interdiscip Toxicol 2:1–12
Wu Q, Chang Q, Wu C, Rao H, Wang Z (2010) Ultrasound-assisted surfactant-enhanced emulsification microextraction for the determination of carbamate pesticides in water samples by high performance liquid chromatography. J Chromatogr A 1217:1773–1778
Yelmaz E, Soylak M (2013) Ionic liquid-linked dual magnetic microextraction of lead(II) from environmental samples prior to its micro-sampling flame atomic absorption spectrometric determination. Talanta 116:882–886
Yongnian N, Qiu P, Kokot S (2005) Simultaneous voltammetric determination of four carbamate pesticides with the use of chemometrics. Anal Chim Acta 537:321–330
Zhang J, Kee Lee H (2006) Application of liquid-phase microextraction and on-column derivatization combined with gas chromatography–mass spectrometry to the determination of carbamate pesticides. J Chromatogr A 1117:31–37
Zhao G, Song S, Wang C, Wu Q, Wang Z (2011) Solid-phase microextraction with a novel graphene-coated fiber coupled with high-performance liquid chromatography for the determination of some carbamates in water samples. Anal Methods 3:2929–2935
Zhou Q, Fang Z (2015) Graphene-modified TiO2 nano tube arrays as an adsorbent in micro-solid phase extraction for determination of carbamate pesticides in water samples. Anal Chim Acta 869:43–49
Acknowledgments
The authors thank the Research Council of the Tabriz University of Medical Science for financial support as grant No. 623110 with ethical code of IR.TBZMED.REC.1396.872.
Funding
Mohammadali Torbati has received research grants from the Tabriz University of Medical Science.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Sanaz Ahmadzadeh Anvar declares that she has no conflict of interest. Mohammadali Torbati declares that he has no conflict of interest. Mir Ali Farajzadeh declares that he has no conflict of interest. Mohammad Reza Afshar Mogaddam declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ahmadzadeh Anvar, S., Torbati, M., Farajzadeh, M.A. et al. Elevated Temperature Homogeneous Liquid Phase Extraction Coupled to Ionic Liquid–Based Dispersive Liquid–Liquid Microextraction Followed by High-Performance Liquid Chromatography: Application of Water-Miscible Ionic Liquids as Extraction Solvent in Determination of Carbamate Pesticides. Food Anal. Methods 13, 1282–1291 (2020). https://doi.org/10.1007/s12161-020-01742-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-020-01742-2