
Abstract
Three sample preparation procedures were compared for a multi-residue analysis of pesticides in hops: (a) modified Hengel’s method based on extraction with acetonitrile in combination with clean-up on a C18 SPE column, (b) miniaturized Biendl’s method based on acetone extraction and PSA SPE sample clean-up, and (c) modified Quick, Easy, Cheap, Effective, Rugged, and Safe (QuEChERS) method which utilizes a specific mixture of three sorbents (PSA, C18, and Z-Sep) for dispersive SPE sample clean-up. The performance of the methods was evaluated and validated for a mixture of 56 pesticides analyzed by the liquid chromatography–high resolution mass spectrometry in compliance with the analytical quality control criteria of the SANTE/11813/2017 guidelines. Strong matrix-dependent signal suppression caused by the co-eluting hop matrix was observed for all sample preparations involved in this method comparison study. The matrix effects in percentages (%ME) were used for the evaluation of the effectiveness of the individual sample clean-up procedures. The recovery experiments were performed by spiking pesticides at the concentration level 0.50 mg/kg into the blank matrix to evaluate the extraction efficiency of the compared methods. Recoveries obtained for the modified Hengel’s method were in the range of 70–120% with RSDs of less than 20% for all studied pesticides. The performances of the methods were tested on the set of 24 samples of hops harvested in the Czech Republic. The method comparison on the determined concentration levels of the pesticide residues clearly showed that the extraction efficiency of the QuEChERS method is significantly less effective for the extraction residues presented in a sample above the level 20 mg/kg. In terms of time consumption, labor, materials, and solvents consumptions, the methods were thoroughly compared, and these demands increase in the following order QuEChERS < modified Hengel’s < miniaturized Biendl’s method.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hops are an essential ingredient in beer production that gives it a typical taste and aroma. Cone flowers of a female hop plant (Humulus lupulus L.) contain resins and oils inside lupulin glands, and these are the main source of various compounds responsible for a bitter flavor and unique aroma. A little-known fact about hop growing is that hop plants are one of the hardest crop choices in farming. Hops require high attention throughout the whole year; however, especially during the growing period, pesticide treatment against various pest common in hop yards (insects, mites, fungal diseases, and weeds) is essential to reach good yields, reduce economic losses, and improve the quality of the final product. In fact, hop plants belong to crops with intensive chemical protection and residues of these agrochemicals are, therefore, widely presented in hops at various concentration levels.
Hops is a very specific and challenging matrix for an analyst because of a high content of co-extracted compounds, such as soft and hard resins, phenolic compounds, chlorophyll pigments, and essential oils. The co-elution of these interfering substances with target analytes causes matrix effects that could dramatically affect analytical performance of a method, especially when LC-MS with electrospray ionization (ESI) technique is applied for sample analysis (Zhou et al. 2017). The significant loss in response (ion suppression) could be, therefore, observed due to the alteration of ionization efficiency of most of target analytes in the presence of co-eluting compounds coming from hop matrix. Thus, it was necessary to develop a clean-up protocol to eliminate undesirable interferences and to avoid negative matrix effects. Hengel and Miller (2008) published the first suitable multi-residue method for the determination of eleven pesticides in dried hops in 2008. The list of analytes was later extended by Hengel (2011) for a wider scope of LC-amenable pesticides. This method is based on acetonitrile extraction in combination with a solid-phase extraction (SPE) column clean-up procedure. Biendl et al. (2014) published a method for the determination of 46 pesticides in hops that is based on extraction by acetone, partitioning with dichloromethane followed by sample clean-up of the crude extract using two interconnected SPE cartridges with silica gel/Florisil sorbents and primary-secondary amine (PSA) for GC-MS and LC-MS analysis, respectively. An original unbuffered QuEChERS sample preparation approach (Anastassiades et al. 2003) with a specially optimized mixture of sorbents for dispersive SPE clean-up has been recently published by Dušek et al. (2018a). This method was optimized for fast high-performance analysis of 48 pesticides. These three methods apply a completely different sample preparation approaches using acetonitrile or acetone as an extraction solvent with or without prior addition of water into the sample, and with or without the salting-out purification procedure. Thus, the additional clean-up steps are also unique for each of these methods.
The aim of this study was to compare three different sample preparation procedures previously published for pesticide residue analysis in hop matrix. These three methods, involving various sample clean-up approaches, have been thoroughly optimized and validated following the SANTE/11813/2017 criteria. Our attention was focused on the ability of each method to effectively minimize co-extraction of matrix components (chlorophyll, resins, bitter acids) which might cause a massive signal suppression, elevated background, and other negative matrix effects. The methods were combined with analysis, and 56 pesticides were involved in this study. The extraction efficiency of each sample preparation procedure, based on the recovery calculation of 56 spiked pesticides, was also evaluated. Moreover, pesticide residue results in field samples, material demands, and laboriousness were also involved in this critical evaluation.
Materials and Methods
Chemicals and Material
Acetonitrile, methanol, formic acid, ammonium formate (all LC-MS grade), acetone, dichloromethane (ASC grade), sodium citrate tribasic dihydrate, and sodium hydrogencitrate sesquihydrate were purchased from Sigma-Aldrich (Steinheim, Germany). Sodium chloride (anal. grade) was obtained from Lach-Ner (Neratovice, Czech Republic). Magnesium sulfate (anal. grade, >98%) was got from Penta (Prague, Czech Republic). Supelclean PSA (50 μm, endcapped, 8.4% carbon load), Discovery DSC-18 (50 μm, endcapped, 18% carbon load), Z-Sep (22 μm), and Supelclean PSA SPE Tube (200 mg/3 mL) were obtained from Supelco (Bellefonte, PA, USA); Strata-X Polymeric Reversed Phase SPE Tube (33 μm, 200 mg/3 mL) was obtained from Phenomenex (Torrance, CA, USA). Ultrapure water was obtained using a Milli-Q system from MilliporeSigma (Burlington, MA, USA) with a resistivity of 18.2 MΩ-cm.
Pesticide standards shown in Table 1 and internal standards (azoxystrobin-d4, thiamethoxam-d3, and triphenyl phosphate) were from Sigma-Aldrich (St. Louis, MO, USA). Standard and internal standard stock solutions (1.0 mg/mL for all pesticides except for 0.2 mg/mL for carbendazim) were prepared by dissolution of neat standards in acetonitrile or, in case of a solubility problem, in methanol or acetone. Working standard mixture, including all 56 pesticides at the concentration of 1 mg/L each, was then prepared by appropriate dilution of the stock solutions with acetonitrile. All solutions were stored at − 20 °C.
Sample Preparation
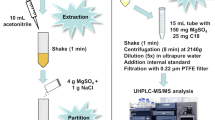
Method B
An amount of 2.0 g (± 0.05 g) of homogenized, previously milled hop sample was weighed into a 50-mL centrifuge tube (recovery samples were fortified at this point) and 100 μL of an internal standard spiking solution (10 mg/L) and 10 mL of water were added. The sample was vortexed for 1 min to let the water soak into the matrix, and then, volume of 20 mL of acetone was added. The tube was tightly capped, and the sample suspension was extracted using a shaking device (260 rpm) for 30 min. Finally, the sample was centrifuged at 5000 rpm for 10 min. Five milliliters of sample extract was then pipetted into a 15-mL centrifuge tube, and 1 mL of a saturated sodium chloride solution and 2 mL of dichloromethane were consequently added. The sample was agitated manually for 1 min, followed by 1 min centrifugation at 2000 rpm for a complete phase separation. After the separation of the organic (≈ 5 mL) and the aqueous layer (≈ 3 mL), a 2-mL aliquot of the upper organic layer was then transferred, using a glass pipet, into a 25-mL pear-shaped flask, evaporated to dryness at 40 °C, and then re-suspended in 2 mL of acetonitrile.
For SPE, a Supelclean PSA column (200 mg/3 mL; Supelco, Bellefonte, PA, USA) was preconditioned with 1 ml of acetonitrile. When the solvent reached the top of the packing, a 1.6-mL aliquot of the sample extract previously suspended in acetonitrile was loaded to the SPE column and eluted with 10 mL of acetonitrile. The eluate was collected into a 25-mL pear-shaped flask and evaporated until dryness using a rotary evaporator at 40 °C. The final extract was diluted with 1 mL of acetonitrile to the final concentration equivalent of 0.1 g matrix per 1 mL and analyzed directly via LC-MS/MS.
Method H
An amount of 1.0 g (± 0.05 g) of a previously homogenized hop sample was weighed into a 50-mL plastic centrifuge tube (recovery samples were fortified at this point), and 50 μL of an internal standard spiking solution (1 μg/mL) and 10 mL of an extraction solution consisting of acetonitrile/water/toluene (80:15:5, v/v) were added. The tube was first vortexed for 1 min and then centrifuged at 4500 rpm for 7 min. For the SPE extraction under the gravity flow, a Strata-X (33 μm, 200 mg/3 mL; Phenomenex, Torrance, CA, USA) cartridge was pre-conditioned using 1 mL of the extraction solvent. When the solvent mixture reached the top of the packing bed, 1 mL of extract was loaded and then eluted with 3 mL of acetonitrile. The flow-through and the wash were collected into a 15-mL centrifuge tube and processed by a second SPE clean-up procedure. For the second SPE procedure, a Supelclean PSA column (200 mg/3 mL) was preconditioned with 3 mL of acetonitrile. When the acetonitrile reached the top of the packing bed, the whole eluate from the first SPE clean-up was loaded in parts on a preconditioned column, and the eluate flowed through the sorbent bed under the gravity flow and was collected into a 25-mL pear-shaped flask. Finally, the 15-mL centrifuge tube was thoroughly rinsed by 3 mL of acetonitrile and the acetonitrile wash was then loaded onto the column. This procedure was repeated twice. The whole eluate was then evaporated to the dryness using a rotary evaporator at 40 °C and resuspended in 1 mL of acetonitrile for subsequent LC-MS analysis.
Method Q
An amount of 1.0 g (± 0.05 g) of well-homogenized, previously milled, hop sample (recovery samples were fortified at this point) was hand-shaken with 10 mL of cold water in a 50-mL polypropylene centrifuge tube for 1 min to hydrate the samples. After 15 min, 10 mL of acetonitrile and 50 μL of internal standard solution (concentration 10 mg/L) were added and the sample was vortexed for 1 min. A pre-weighted salt mixture containing a blend of 4 g MgSO4, 1 g NaCl, 1 g trisodium citrate, and 0.5 g disodium hydrogenate sesquihydrate was dispensed into the tube. The tube was first immediately shaken by hand for 1 min to prevent the formation of crystalline agglomerates and then centrifuged at 4500 rpm for 7 min. Six milliliters of supernatant were transferred into a 15-mL polypropylene centrifuge tube containing 900 mg of anhydrous MgSO4. The tube was sealed and vortexed for 30 s. The sample was then centrifuged at 5000 rpm for 5 min. An aliquot of 1 mL of supernatant was transferred into a 2-mL screw cap microtube containing a blend of 100 mg PSA, 25 mg C18, and 25 mg Z-Sep dispersive SPE sorbents. The tube was carefully sealed, vortexed for 30 s, and then centrifuged at 13,000 rpm for 5 min. Finally, 800 μL of supernatants were carefully transferred into a vial and subsequently analyzed via LC-MS/MS.
LC-MS/MS Analysis
The LC separation was carried out using a Dionex UltiMate 3000 UHPLC system (Thermo Scientific, Waltham, MA, USA). Analytes were separated on a reversed-phase C18 Atlantis T3 column (2.1 × 100 mm, 3 μm) from Waters (Milford, MA, USA) with a guard C18 column (SecurityGuard ULTRA) from Phenomenex (Aschaffenburg, Germany). A gradient of a mobile phase A (water containing 2 mM ammonium formate and 0.1% formic acid) and a mobile phase B (methanol) supplied at 0.34 mL per minute was applied. The initial mobile phase started from 85:15 (v/v) of mobile phase A, and the program consisted of 0–0.5 min at 85:15; from 0.5 to 9 min, the mobile phase was linearly increased to 5:95; and from 9 to 15 min held at 5:95. The gradient was then returned to the initial conditions in 0.1 min and held there for 2.9 min for stabilization. The column oven was heated to 40 °C, and the injection volume was 2 μL.
The Q-Exactive hybrid quadrupole-orbitrap mass spectrometer (Thermo Scientific, Waltham, MA, USA) was operated in the positive electrospray ionization (ESI) mode acquiring two high-resolution transitions for each analyte in a parallel reaction monitoring (PRM) data acquisition mode. The precursor ions from the scheduled inclusion list were isolated and fragmented within the retention time window of ± 0.3 min, filtered in the quadrupole at isolation window (target m/z ± 0.7 amu), and fragmented in an HCD collision cell, and product ions were collected in the C-trap at 17,500 resolution (FWHM, full width at half maximum, at m/z 200), the AGC target value of 2e5, and maximum ion injection time of 40 ms. The instrument was externally calibrated prior to each measurement using the mixture of mass calibrants. Data processing was performed using TraceFinder software version 4.1 (Thermo Scientific).
The parameters pertaining to the PRM transitions were developed and optimized by infusion of standard solutions prepared in the mobile phase mixture (50/50) with a syringe pump. To determine optimum PRM transitions, the high-resolution MS/MS spectrum of each compound was inspected for two specific fragment ions selected for quantification (Q2) and confirmation (Q3) purposes in the quantitative assay. Finally, unique pairs of PRM transitions were monitored per analyte. The normalized collision energy (NCE) was individually optimized for each pair of PRM transitions. The PRM data acquisition mode operates with parallel detection of Q2 and Q3 transitions in a single analysis. Therefore, the NCE was optimized to strike a balance between the maximum response of the quantification ion and the sufficient intensity of the confirmation ion. The ratio of the quantification and confirmation ions was used as an identification criterion. The maximum allowable deviation from the expected ion ratio (calculated from standard solutions) of 30% for unknown samples was applied in accordance with the criteria stated in the EU guidance document SANTE/11813/2017. The limit of detection (LOD) and the limit of quantification (LOQ) were calculated using the equations LOD = 3Sy/x ÷ b and LOQ = 10Sy/x ÷ b, where b is the slope of the calibration curve and Sy/x is the standard error of regression (Stone and Ellis 2011). The retention time, mass of precursor ion (Q1, m/z), observed (experimental) masses of two product ions (Q2 and Q3, m/z), NCE, and LOQ for each compound of interest are shown in Table 1.
Quantification
An external matrix-matched calibration method was used, and concentrations of pesticides were determined by interpolation of relative peak areas of each pesticide to an internal standard peak area in the sample. In order to compensate for losses during sample processing and instrumental analysis, internal standards (azoxystrobine-d3, thiamethoxame-d4, and triphenyl phosphate) were used. Corresponding isotopically labeled internal standards were used for azoxystrobin and thiamethoxam; triphenyl phosphate (TPP) was used as an internal standard for all remaining analytes. Quantitation was performed through 5-point matrix-matched calibrations prepared in the range of 0.010–0.200 mg/L by addition of an appropriate volume of the pesticide working standard mixture and the internal standards at a concentration level of 0.5 mg/kg to the extracts of the organically grown hops. One milliliter of the blank sample, prepared under the sample preparation protocols of the abovementioned methods H, B, and Q, was evaporated to the dryness, and appropriate volumes of solvents were added prior to the solvent reconstitution with acetonitrile to make the final volume of 1 mL.
If the content of a sample was not within the range of calibration curves, quantitation would be done using a standard addition method. The diluted acetonitrile extract of each hop sample (50 or 100 μL each) was pipetted into four 2-mL glass vials, three of which were fortified with the pesticide working standard mixture at a concentration of 1 mg/L to make the standard addition calibration curve corresponding to 50, 100, and 150% of expected concentration of pesticide residues in the sample extract. The appropriate volume of acetonitrile was added to each vial to make the final volume of 600 μL or 1 mL, respectively.
Methods Validation
The validation study was performed based on the European SANTE/11813 guidelines (2017). Performance characteristics of each individual method obtained via the validation process are summarized in Tables 1 and 2. In order to evaluate the accuracy (expressed as recovery) and precision (repeatability, expressed as a coefficient of variation in percentages) of all three methods, recovery experiments were performed. A blank sample of organically grown hops was spiked with a known amount of a pesticide mixture solution (1 mg/L) to obtain a concentration of all analytes at 0.50 mg/kg level. The spiking level was chosen to be at least ten times higher than LOQs and below the current maximum residue limits (MRL) as well.
The limits of quantification (LOQ) were estimated for the methods H, B, and Q by dilution of pesticide standard mixture with the blank sample extracts prepared under the sample preparation protocols to the expected LOQ, and recovery and precision were determined using these samples (n = 5). A precision (RSD, %) of no more than 20% of variation was considered to be acceptable. The LOQ values for target analytes were in the range of 0.02–0.05 mg/kg (see Table 1). The linearity of the developed method was tested over the concentration range of 0.010 to 0.200 mg/L by a matrix-matched calibration curves. The calculated regression coefficient (r) values were found to be > 0.999 for most of the target analytes.
Results and Discussion
Optimization of Extraction Procedures
Sample preparation methods for the analysis of pesticide residues in hop matrix published by Hengel (Hengel 2011) and Biendl (Biendl et al. 2014) had to be modified and optimized before the beginning of the method comparison study. As the first step of the sample clean-up efficiency comparison, matrix effects of individual methods were evaluated. To make this possible, it was essential to modify the sample preparation procedures in the way that the matrix concentration per 1 mL of the final extract was the same for all evaluated methods. Therefore, the sample preparation procedures B and H were modified to achieve the matrix concentration 0.1 g sample per milliliter of the final extracts. Practically, it was not possible to use more than 2 g of a sample and 10 mL of water as complete water absorption by the sample matrix was necessary. The method designed by Biendl et al. (2014) was mainly modified to reduce organic solvent consumption needed for the sample preparation. The solvent requirements needed for the sample preparation by method B were finally reduced to 20 mL of acetone, 2 mL dichloromethane, and acetonitrile compared to 100 mL acetone, 10 mL dichloromethane, and 10 mL of ethyl acetate previously used in the original method. Besides the reduction of solvent demands, the matrix concentration in the final acetonitrile extract was changed from 0.125 to 0.1 g sample/mL. The principle of the sample preparation procedure published by Biendl et al. (2014) was not changed, and method B, evaluated in this study, newly presents the miniaturization of the whole procedure that led to five times lower solvent consumption.
The method published by Hengel (2011) had to be also modified for the purpose of this method comparison because the hop matrix concentration was originally only 0.02 g in 1 mL of the final extract. Previously presented results (Dušek et al. 2018b) clearly showed that matrix effects of the Hengel method for most evaluated pesticides tend to be higher by approximately 15% in comparison with the QuEChERS method (method Q), even though the matrix concentration per milliliter is five times lower. In the first step of the Hengel method modification, the extraction solvent was changed. Especially in case of dry samples, the extraction solvent has to have a high dissolving ability for pesticides and good permeability into the matrix (Kolberg et al. 2011). Both Hengel’s and QuEChERS method use acetonitrile as an extraction solvent. Nevertheless, the original QuEChERS method was, for matrixes with water content lower than 25%, modified and water was added to the sample prior to the extraction to achieve maximum extraction yield and accurate results (Čajka et al. 2012). Therefore, in the first step, acetonitrile for extraction was replaced by the mixture of acetonitrile–water–toluene (80/15/5). Toluene was added into the extraction mixture to compensate reduced extraction efficiency of acetonitrile-water mixture for non-polar compounds. The mixture of three solvents (sorted in order of increasing polarity index (Harris 1998), PI): toluene (PI = 2.4), acetonitrile (PI = 5.8), and water (PI = 10.2), was finally used for the extraction of pesticides with various polarity. Originally, Hengel method used a solid-phase extraction (SPE) procedure for removal of mainly lipophilic compounds such as co-extracted resins and residues of aromatic hop oils. However, the water presented in the extraction solvent is, in this case, able to increase the retention of these compounds during reversed-phase SPE clean-up. Apart from a wide range of nonpolar compounds, acetonitrile or acetone hop extract contains mainly various analogues of hop bitter acid (α- and β-bitter acids) that forms a majority of extractable compounds of hops matrix (Belitz et al. 2009). The primary-secondary amine (PSA) sorbent could effectively remove these compounds together with other interfering compounds such as sugars, fatty acids, organic acids, and anthocyanin pigments (Schenck and Lehotay 2000; Schenck et al. 2002). Therefore, the SPE procedure using the PSA cartridge was used for additional clean-up of the final acetonitrile extract. The efficiencies of the original and modified preparation procedures were compared evaluating the ability to eliminate matrix suppression. The method B showed matrix effects (%ME) for most evaluated pesticides about 10 to 30% lower compared to the original Hengel method. The proposed modification of Hengel method, method B, allows efficient pesticide extraction without any salting-out clean-up step and is applicable and rugged for selected pesticide analysis in hops at the screening level that is approximately five times lower than the original Hengel method.
Comparison of Cleaning Efficiency—Matrix Effects
The influence of matrix effects was used to assess the effectiveness of a clean-up procedure of each sample preparation. Matrix effects (ME) in percentage were calculated for all compound–sample preparation procedure combinations (H, B, and Q) comparing slopes of calibration curves created with solvent standard and matrix-matched standards. The following equation was used: ME (%) = (slope matrix ÷ slope solvent) × 100. Five-point calibration curves from 0.010 to 0.200 mg/L were prepared in solvent and in extracts of the pesticide-free hops. The linearity of the calibration curves (expressed as r2) was also evaluated and, in all cases, good linearity was achieved with correlation coefficients > 0.997. As can be seen in Table 2, most analytes showed negative values when evaluating the matrix effect. Generally, the negative values represent signal suppression in hop matrix compared to the solvent. Values lower than 20% indicate no or rather insignificant matrix effects. Values from 20 to 50% correspond to medium matrix effects, and values higher than 50% are considered as strong matrix effects. For 56 analyzed pesticides, methods H, B, and Q showed strong matrix effects for 37, 36, and 40 compounds and medium matrix effects for 13, 10, and 16 compounds, respectively. It seems that all applied sample preparation methods have comparable efficiency in terms of a sample clean-up. In addition to that, the ability of each method to effectively remove the co-extracted matrix components is compared through the scatter plots. Figure 1a–c shows plots of calculated matrix effects for 56 pesticides for one sample preparation method in comparison with the others and their linear fit. The satisfactory correlation coefficients (r) 0.91, 0.93, and 0.96 were found for the scatter plots of method H vs. method B (see Fig. 1a), method Q vs. method H (see Fig. 1b), and method Qvs. method B (see Fig. 1c), respectively. The figures could be also interpreted using the following rule: if the data point is below the 1:1 ideal regression line, then the method with the values plotted on the x-axis has lower matrix effect, and higher matrix effect has the method with the values plotted on the y-axis. Figure 1a shows the comparison between the method H and B. It can be seen that most data points are close to the line of equity, and therefore, these two methods provide the sample clean-up with the same effectiveness. The comparisons between methods H and Q (see Fig. 1b) showed that there is no observable difference between these two methods for compounds with strong matrix effects but compounds with matrix effects lower than 60% were significantly less affected by co-extracted matrix components when method H was applied. This was also observed in the case of method B and Q comparison (see Fig. 1c). The intercept values of the regression curves of the scatter plots comparing the matrix effects of the methods H and Q, and methods B and Q were 20.065 and 19.241, respectively. It indicates similar effectiveness of the sample clean-up using method Q in comparison with methods H and B. In addition to that, Figure 1c also shows that almost all data points are above the line of equity (x = y line) that indicates that for majority of 56 analyzed compounds, method B is generally more efficient than method Q. Table 2 summarized calculated matrix effects (%ME) for each compound–sample preparation procedure ordered by increasing retention time. It is obvious that various sample preparation procedures affected mainly early eluting compounds for which most significant differences among matrix effects values were observed. However, at the same time, all methods produce samples that show strong signal suppression at the end of the chromatography. Strong matrix effects at the end of a reverse-phase chromatographic analysis indicate co-extraction of hydrophobic compounds that none of these methods was able to fully remove.

Scatter plots with regression line comparing the matrix effects (%ME) of a methods B and H, b methods Q and H, and c methods Q and B. Solid red line represents the line of equity (y = x line)
Evaluation of Methods’ Recoveries
The percent recovery values for 56 pesticides and each sample preparation procedure were calculated using five replicates. The average recoveries (%) for methods H, B, and Q at spiking level 0.50 mg/kg were 103 ± 9, 89 ± 18, and 82 ± 19, respectively. The recovery data summarized in Table 2 show that method H gives recovery within the acceptable interval of 70–120% defined by SANTE document and with precision (RSD) lower than 20% for almost all evaluated pesticides except for spirotetramat. The other two methods, B and Q, provided recovery results for 4 and 8 pesticides outside the acceptable range of 70–120%, respectively. These two methods, on the contrary to the method H, involve in the sample preparation procedures of the salt-induced liquid–liquid extraction (LLE) step, which has a strong negative effect on the extraction yield mainly for highly polar pesticides. Polar pesticides such as acephate, pymetrozin, and propamocarb are highly soluble in water, and therefore tend to stay in the water phase during salting-out of the organic solvent (acetone–water or acetonitrile–water extraction solutions). The recoveries of acephate, pymetrozin, and propamocarb extracted by methods B and Q were 13% and 45%, 13% and 7%, 34% and 9%, respectively. The recovery lower than 70% could pass the acceptance criteria (SANTE/11813/2017) for validation only if (1) the recovery rate is consistent, (2) the RSD value is lower than or equal to 20%, and (3) the mean recovery value is not lower than 30%. It is required that the analytical results are, in this case, corrected for recovery, or more accurate method should be used, if available. For example, propamocarb is considered to be a non-QuEChERS-amenable pesticide. A method, known as a QuPPe method (Anastassiades et al. 2015), which is based on the extraction of an acidified sample by a methanol–water solution could be successfully used for the determination of propamocarb and other highly polar pesticides. The recovery rate of pymetrozin, which is pH-dependent, could be well above 70% if the pH value is increased during the extraction from pH value around 4 to pH above 5. The pH value change causes the replacement of the citrate salts with Na-acetate or Mg-acetate salts (EURL-SRM Analytical Observation Report 2016). As these two examples show, alternative approaches or modifications to the method Q that have acceptable recovery for these two compounds are available.
The recovery rates of problematic highly polar pesticides such as acephate, pymetrozin, and propamocarb demonstrated the main advantage of the method H that meets the method performance acceptability criteria, accuracy (recovery), and repeatability, for all 56 evaluated compounds. Potentially, the scope of the analytes included in the method H could be broadened to include other highly polar, non-QuEChERS-amenable, pesticides. The method B gives unsatisfactory recovery results for acephate and pymetrozin and the method Q for pymetrozin and propamocarb. On the other hand, it should be noted that from these three compounds, only insecticide pymetrozin is approved for treatment, and moreover, its routine application against hop aphids is quite rare. The list of pesticides approved for hop treatment is limited, and for example, from 56 pesticides involved in this study, only 18 pesticides are allowed for the application on hops in the Czech Republic. The data summarized in Table 2 compare the recoveries of the methods, and it is clearly visible that there are no significant differences among recoveries of the pesticides approved for hop treatment (highlighted in italics) for these three sample preparation methods. The method H definitely seems to be the most universal multi-residue method applicable for a wide range of analytes in comparison with the other two methods. Nevertheless, in practice, all three methods H, B, and Q are applicable for the determination of pesticides that are approved and routinely used for hop treatment.
Method Comparison Based on Pesticide Results in Field Samples
The matrix spike recovery results showed, with a few exceptions, that the methods B, H, and Q provided recovery values near to 100 ± 20% for the fortification level 0.50 mg/kg (see Table 2) with RSD < 20%. Therefore, these methods could be considered as multi-residue methods suitable for the determination of pesticides in hops. In the next step, 24 samples of hops were analyzed for pesticide residues by all three methods to check and verify their performance on the set of field samples. The residues of fungicides, such as mandipropamid, boscalid, azoxystrobin, pyraclostrobin, dimethomorph, and ametoctradin, were detected above LOQ in 23, 15, 13, 13, 12, and 11 samples, respectively. The residues of insecticides spirotetramat and fenpyroximate were detected above LOQ values in 11 and 5 samples, respectively. The individual concentrations in samples varied up to three orders of magnitude. For example, the residues of mandipropamid were found at concentrations ranging from 0.075 to 31 mg/kg. In total, 122 individual pesticide residues were reliably quantified in the samples, and these data were used for the construction of the scatter plots (see Figure 2a–c) comparing the residue results of each method–method pair. Figure 2a shows the scatter plot illustrating the relationship between the residue results of the method H and B in 24 hop samples. The scatter points, especially the scatters for the concentrations lower than 15 mg/kg, lie tightly to the solid red line, which represents the equity line with the correlation coefficient r = 0.992 and slope value 0.9193. The slope value is lower than 1, which is the ideal slope value of the equity line, and indicates that the results of one method are lower than of the other ones. This is in accordance with the scatter plot in Fig. 2a that clearly shows that the method H (x line) gives a bit higher results than method B (y line), especially for concentrations higher than 20 mg/kg. The results of ametoctradin were, for instance, 52 and 46 mg/kg, 37 and 32 mg/kg, or 29 and 25 mg/kg in a pair of samples determined by the methods H and B, respectively. Nevertheless, the results indicate that these two sample preparation procedures, method H and B, are comparable in terms of pesticide determination in hop samples because no significant biases have been noted between them. The other two plots, Fig. 2b and c, show the results of the method H and B plotted against results of the method Q, respectively. The scatter points in these figures appear to be less grouped along the equity line compared to the scatter points in Fig. 2a, again especially for the residues higher than 20 mg/kg. Moreover, the data shows that mainly the scatter points representing results of the ametoctradin residues lie far above the equity line. Except the scatter plots of ametoctradin, results of the method Q are more or less comparable with the results of the methods H and B. This is indicated by the acceptable correlation coefficients r > 0.9 and the slope values 1.112 and 1.042, respectively, which are relatively close to the ideal slope value (slope = 1) of the equity line. Overall, from the method comparison based on the results of all pesticide residues in field samples, it could be concluded that the significant differences were primarily observed for the residues at the concentrations above 20 mg/kg and that the method B and Q showed significantly lower results of fungicide ametoctradin than the method H. The scatter plots for ametoctradin (Fig. 3a–c) and pyraclostrobin (Fig. 4a–c) were also constructed. Figure 3a shows that the scatters lay along the equity up to the concentration of 30 mg/kg and that the results of the method B tended to be lower in comparison with the method H. On the contrary, the scatter plots on Fig. 3b and c clearly show that all scatters lay above the equity line on both graphs that indicate that the method Q gives systematically lower results compared to the methods H and B. The percentage recovery of ametoctradin in spiked hops was 68 ± 2 (see Table 1). This value, which is on the edge of the acceptable interval defined by the SANTE document (SANTE 11813 2017), clearly indicates low extraction efficiency of the acetonitrile/water mixture (50/50, v/v) compared to the acetone/water mixture (67/33, v/v) used in the method B or the extraction mixture of acetonitrile/water/toluene (80/15/5, v/v) as used in the method H. Ametoctradin is a strong acid (pKa = 2.78) with the octanol–water partition coefficient log P = 4.4 (PPDB 2007). It is relatively nonpolar, and hence, the least polar solvent should be required for its full extraction from the matrix. A mixture of organic solvents with different polarities is, therefore, beneficial to use for an effective extraction of a wide range of analytes, especially from hop matrix. Although ametoctradin was not involved in the original study published by Hengel (2011), uses acetonitrile as well; however, in this case, only 0.5 g of hops is extracted by 15 mL of net acetonitrile. Although ametoctradin was not involved in the original study published by Hengel (2011), the data presented by Dušek et al. (2018b) showed the 97% recovery of ametoctradin spiked at the level of 1.0 mg/kg. Almost ideal recovery was in this case achieved most probably due to a relatively small sample dose and its extraction by net acetonitrile without any water addition. Figure 4a–c shows the scatter plots for pyraclostrobin, a fungicide found in 13 from 24 hop samples. The method comparison for pyraclostrobin clearly shows that there are no significant differences between the residue results obtained by all tested methods. The extraction effectivity is almost the same in the whole concentration range up to 2 mg/kg. Moreover, the comparison of the residue results of methods B and Q shows an excellent agreement between them (see Fig. 4c). Therefore, the selection of a proper extraction solvent is a crucial step in the method optimization. The method comparison data proved that all tested methods have up to approx. 20 mg/kg, the same effectiveness of the pesticide residue extraction (except for fungicide ametoctradin). On the contrary, the pesticide residues could be found quite often in hops at the levels above this concentration, and in this case, especially the method Q tends to be less efficient for the extraction of pesticide residues.

Scatter plots with regression line of pesticide residues (ametoctradin, azoxystrobin, boscalid, dimethomorph, mandipropamid, fenpyroximate, pyraclostrobin, and spirotetramat, (n = 112) found in 24 samples of hops prepared by a methods B and H, b methods Q and H, and c methods Q and B. Solid red line represents the line of equity

Scatter plots with regression line of ametoctradin residues (n = 11) found in 24 samples of hops prepared by a methods B and H, b methods Q and H, and c methods Q and B. Solid red line represents the line of equity

Scatter plots with regression line of pyraclostrobin residues (n = 13) found in 24 samples of hops prepared by a methods B and H, b method Q and H, and c methods Q and B. Solid red line represents the line of equity
Comparison of Methods H, B, and Q with Regard to Labor
This method comparison study evaluated numbers of different steps involved in the sample handling protocol of three different sample preparation methods and as well as the requirements on chemicals, solvents, and material. Table 3 clearly summarizes the steps of each preparation procedure previously described in the literature and also their modifications (method H, B, and Q) involved in this study. Evaporation of the sample eluate using a rotary evaporator is definitely the most time-consuming step of the whole procedure. From this point of view, the most time-consuming method is the method B (mini-Biendl extraction method), as it involves solvent evaporation to the dryness prior and after the sample clean-up step on SPE column. The modified QuEChERS sample preparation approach (method Q), on the contrary, does not involve a solvent exchange step and any SPE column clean-up procedure, and thus, it could be considered as the fastest and least laborious method.
Conclusion
In this paper, two sample preparation procedures originally published by Hengel and Biendl for determination of pesticide residues in hops were modified and miniaturized. The two methods, H and B, and the QuEChERS-d-SPE sample preparation procedure (method Q) were compared systematically through the evaluation of recoveries (extraction efficiency) and matrix effects (the effectiveness of the clean-up approach). The extraction procedures were evaluated for the extraction of 56 target compounds covering pesticides frequently applied on hops determined by LC-HR-MS/MS method. Mean recoveries (n = 5) were between 70 and 120% with relative standard deviations (RSD) lower than 20% in most of the cases except acephate, pymetrozine, and propamocarb which showed dissatisfactory recoveries for method B and Q. Moreover, the application of primary secondary amine (PSA) as a SPE sorbent negatively affects the recovery of compounds with acidic functional compounds such as flonicamid metabolites TFNA and TFNG. The matrix effects of the proposed methods showed that all sample preparation procedures reduce the influence of the co-extracted matrix compounds more or less in the same effective way, and no significant differences between method B and H were observed. The methods were successfully applied for multi-residue analysis of pesticides in 24 field samples of cone hops; six fungicides (ametoctradin, azoxystrobin, boscalid, dimethomorph, mandipropamid, and pyraclostrobin) and two insecticides (spirotetramat and fenpyroximate) were found in the range of 75 μg to 31 mg per kilogram. The pesticide residue levels determined by each method were compared, and the results showed that all methods generally provide comparable results to one another. Nevertheless, the comparison of ametoctradin residues in field samples clearly showed that method Q gives systematically lower results of this fungicide in the field samples, it is in relation with relatively low recovery of ametoctradin (≈ 70%) in the samples prepared by the method Q. All procedures for sample preparation were also compared on the basis of the different laboratory relevant characteristics like time, materials, and solvent consumptions.
In summary, the results showed that the three different sample preparation procedures, methods H, B, and Q, provide almost the same effective clean-up for analysis of pesticide residues in hop matrix. Nevertheless, each of these methods has specific pros and cons that should be taken into consideration prior to a method selection. The method published by Hengel, method H, is generally applicable for the widest range of analysts; miniaturized Biendl’s method (B) seemed to be the most robust; and method Q, based on QuEChERS sample preparation approach, has definitely less labor compared to other methods.
References
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86(2):412–431
Anastassiades M, Kolberg DI, Eichhorn E, Benkenstein A, Lukačević S, Mack, D, Wildgrube C, Sigalov I, Dörk D, Barth A (2015) Quick method for the analysis of numerous highly polar pesticides in foods of plant origin via LC-MS/MS involving simultaneous extraction with methanol (QuPPe-Method). EU Reference Laboratory for pesticides requiring Single Residue methods (EURL-SRM). http://www.crl-pesticides.eu/library/docs/srm/meth_QuPPe.pdf. Accessed Dec 2018
Belitz HD, Grosch W, Schieberle P (2009) Alcoholic beverages in: food chemistry, 4th edn. Springer Science & Business Media, Berlin, pp 895–896
Biendl M, Jaeger G, Kittsteiner-Eberle R, Schmidt C (2014) Rapid and sensitive determination of pesticide residues in hops and hop products using HPLC-MS/MS and GC-MS/MS. BrSc 67:108–115
Čajka T, Sandy C, Bachanová V, Drábová L, Kalachová K, Pulkrabová J, Hajlšová J (2012) Streamlining sample preparation and gas chromatography–tandem mass spectrometry analysis of multiple pesticide residues in tea. Anal Chim Acta 743:51–60 https://doi.org/10.1016/j.aca.2012.06.051
Dušek M, Jandovská V, Olšovská J (2018a) Analysis of multiresidue pesticides in driedhops by LC–MS/MS using QuEChERS extraction together with dSPE clean-up. J I Brewing 124:222–229 https://doi.org/10.1002/jib.490
Dušek M, Jandovská V, Olšovská J (2018b) Pesticide residue analysis in hops: four different procedures of sample preparation are currently used. Is one of them more useful than the others? 55th North American Chemical Residue Workshop [Online], Naples, FL, USA, July 22–25, 2018. Research Institute of Brewing and Malting. https://beerresearch.cz/wp-content/uploads/2019/02/NACRW2018_Poster59_Dusek.pdf. Accessed Feb 2019
EURL-SRM (2016) Analytical observations report analysis of pymetrozine by the QuEChERS method - impact of pH on recovery rate. EU Reference Laboratory for pesticides requiring Single Residue methods (EURL-SRM). http://www.crl-pesticides.eu/userfiles/file/EurlSRM/EurlSrm_Observations_Pymetrozine.pdf. Accessed Dec 2018
Harris D (1998) Quantitative chemical analysis. In: Katz E, Eksteen R, Schoenmakers P, Miller N (eds) Handbook of HPLC, 9th edn. Marcel Dekker Inc., New York
Hengel MJ, Miller M (2008) Analysis of pesticides in dried hops by liquid chromatography-tandem mass spectrometry. J Agric Food Chem 56(16):6851–6856
Hengel MJ (2011) Expanded method development for the determination of pesticides in dried hops by liquid chromatography with tandem mass spectrometry. J Am Soc Brew Chem 69(3):121–126. https://doi.org/10.1094/ASBCJ-2011-0519-01
Kolberg DI, Prestes OD, Adaime MB, Zanella R (2011) Development of a fast multiresidue method for the determination of pesticides in dry samples (wheat grains, flour and bran) using QuEChERS based method and GC-MS. Food Chem 125(4):1436–1442. https://doi.org/10.1016/j.foodchem.2010.10.041
PPDB: Pesticide Properties DataBase (2007) Agriculture & Environment Research Unit (AERU) at the University of Hertfordshire. https://sitem.herts.ac.uk/aeru/ppdb/en/index.htm. Accessed Dec 2018
SANTE/11813 (2017) Guidance document on analytical quality control and method validation procedures for pesticide residues and analysis in food and feed (implemented 01/01/2018. European Commission, Health and Consumer Protection Directorate General. https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2017-11813.pdf. Accessed Dec 2018
Schenck FJ, Lehotay SJ (2000) Does further clean-up reduce the matrix enhancement effect in gas chromatographic analysis of pesticide residues in food? J. Chromatogr A 868(1):51–61
Schenck FJ, Lehotay SJ, Vega V (2002) Comparison of solid-phase extraction sorbents for cleanup in pesticide residue analysis of fresh fruits and vegetables. J Sep Sci 25(14):883–890
Stone DC, Ellis J (2011) Stats tutorial - instrumental analysis and calibration. Department of Chemistry, University of Toronto. http://www.chem.utoronto.ca/coursenotes/analsci/stats/index.html. Accessed Dec 2018
Zhou W, Yang S, Wang PB (2017) Matrix effects and application of matrix effect factor. Bioanalysis 9(23):1839–1844. https://doi.org/10.4155/bio-2017-0214
Funding
The study was supported by the Ministry of Agriculture of the Czech Republic within Institutional Support MZE-RO1918 and by the Charles University within project SVV260440.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Martin Dušek declares that he has no conflict of interest. Vladimíra Jandovská declares that she has no conflict of interest. Kamila Kalachová declares that she has no conflict of interest. Jana Olšovská declares that she has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dušek, M., Jandovská, V., Kalachová, K. et al. Comparative Study of Three Sample Preparation Methods for Multi-residue Extraction of Pesticide Residues in Hop Samples. Food Anal. Methods 13, 503–515 (2020). https://doi.org/10.1007/s12161-019-01658-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-019-01658-6