
Abstract
A highly selective method was established to study the dynamic of biogenic amine formation during the brewing process. The presence of histamine, tyramine, cadaverine, putrescine, phenylethylamine, spermidine, and spermine was determined in raw materials (barley, hop, yeast), intermediate products (sweet wort, hopped wort, green and matured beer), and beers. Dansylated amines were separated on a Zorbax-XDB C18 column using a binary mobile phase composed of acetonitrile and water. Chromatographic conditions were optimized using an experimental design, and validation was performed following International Conference on Harmonization (ICH) recommendations. Calibration data fitted a linear regression model with R 2 > 0.99. Repeatability (n = 6) and intermediate precision (n = 3) in matrix showed relative standard deviation (RSD) values lower than 7.62 and 8.51 %, respectively. Recoveries, at three different concentrations, ranged from 73.97 to 114.94 %. The proposed method was applied to determine the dynamic of biogenic amine content during brewing process of three microbreweries, allowing the detection of the principal sources or stages of biogenic amine (BA) contribution/formation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biogenic amines (BAs) are well-known organic nitrogenous compounds, naturally synthetized in animals, plants, and microorganisms. These amines are generally formed by decarboxylation of free amino acids or by amination or transamination of aldehydes and ketones. BAs are essential for several physiological functions such as body temperature regulation and gastric acid secretion. However, the consumption of foods containing large amounts of BA may cause several adverse symptoms, e.g., headache, hypotension or hypertension, nausea, cardiac palpitations, hot flushes, and respiratory discomfort (Alvarez and Moreno-Arribas 2014; Anli and Bayram 2009). BA occurrence has been reported in several types of foods, e.g., cheeses, fish, vegetables, meats, wines, and beers (Onal et al. 2013; Spano et al. 2010). Their presence is related with food deterioration and contamination, which is associated with sanitary-type deficiencies during production process (EFSA 2011; Kalač and Křížek 2003). In the case of brewing, a general overview classifies BA into two groups or origin sources (Kalač and Křížek 2003); histamine, tyramine, and cadaverine are associated with microbial contamination, principally bacterial, where Pediococcus and Lactobacillus are the main producer species (Almeida et al. 2012). However, some enterobacteria and strains of Saccharomyces could also contribute to BA formation (Bokulich and Bamforth 2013). The second group represented by spermidine, phenylethylamine, agmatine, and spermine is considered “natural” because it comes from raw materials such as malted barleys, yeasts, and hops (Kalač and Křížek 2003). Putrescine presents a dual origin, contributed by raw materials and also produced by bacterial contamination (Loret et al. 2005). Although this general categorization, fermentation process as well as storage are the critical steps that regulate the type and amount of BA. Occurrence of BA in beers has been reported in European (Kalač and Křížek 2003) and Asian countries (Choi et al. 2012; Deetae et al. 2013). In South America, the BA content in beers has only been reported in Brazil (Gloria and Izquierdo-Pulido 1999) and Venezuela (Camacho et al. 2007). From an analytical point of view, BA determination in beers and brewing is a difficult task for several reasons: (i) different kinds of matrices (barley, hop, yeast, beer, etc.), (ii) commonly found at trace levels, and (iii) the lack of relevant chromophore groups on their chemical structures. As indicated above, the brewing requires the combination of diverse matrices, which forces the use of different sample preparation methods for BA evaluation. Extractions with perchloric acid (Kalac et al. 1997), hydrochloric acid/3,3′-thiodipropionic acid (TDPA) mixture (Romero et al. 2003), and toluene (Fernandes et al. 2001) have been reported as well as solid-phase extraction (SPE) using C18 columns (Loukou and Zotou 2003) and cation exchange resins (Halasz et al. 1999). Beer matrix requires a simpler sample preparation that consists in degassing, centrifugation, and filtration (Loukou and Zotou 2003). After extraction, due to the lack of important chromophore groups that allow a satisfactory detection by visible/UV absorption or fluorescence (FLD), BAs are usually derivatized using different reagents, e.g., o-phthalaldehyde (Loret et al. 2005), benzoyl chloride (Aflaki et al. 2014), ninhydrin (Halasz et al. 1999), and dansyl chloride. The latter is the most used because the derivate exhibits a higher stability (Anli et al. 2006; Loukou and Zotou 2003). Regarding separation and quantification techniques, several analytical methods have been applied including high-performance liquid chromatography (HPLC) (Onal et al. 2013), high-performance thin-layer chromatographic (HPTLC) (Kalač and Křížek 2003; Romano et al. 2012), gas chromatography (GC) (Almeida et al. 2012), capillary electrophoresis (Cortacero-Ramirez et al. 2007; Daniel et al. 2015), and sensors (Pospiskova et al. 2013; Ramon-Marquez et al. 2016). HPLC coupled to UV, FLD, and/or MS detectors is the current technique of choice due to its efficiency and detection capability (Choi et al. 2012). Besides the toxicological importance, BA determination is also relevant from a technological point of view. The identification of the principal sources or stages of BA contribution/formation during production process will allow to carry out corrective actions that can reduce or prevent their presence (European Community 2011). Because, even when there are not worldwide official limits for BA content in beers, some regional limits could be a problem for companies that would like to export. Thus, analytical methodologies capable of evaluating the contribution of each raw material and productive step are required. From the few methods published about BA analysis during brewing (Cortacero-Ramirez et al. 2007; Halasz et al. 1999; Izquierdo-Pulido et al. 1994; Kalac et al. 1997; Romero et al. 2003), just Kalac et al. (1997) reported the evaluation of all raw materials, i.e., malted barleys, hops, and yeasts. But, this methodology, using benzoyl chloride, required two parallel analyses to analyze only five BA, one for histamine and tyramine and other for tryptamine, cadaverine, and putrescine evaluation. Following our previous studies about BA determination in wines (Henríquez-Aedo et al. 2012; Pineda et al. 2012), the present work reports an optimized and validated chromatographic method to determine the dynamic of BA content during the entire brewing process including all raw materials. Applying the proposed highly selective method (regardless of matrix), it was possible to evaluate the presence of histamine (His), tyramine (Tyr), cadaverine (Cad), putrescine (Put), phenylethylamine (Phe), spermidine (Spd), and spermine (Spm) in raw materials (barley, hops, yeast), intermediate products (sweet wort, hopped wort, green and matured beer), and beers.
Material and Methods
Regents and Samples
Putrescine dihydrochloride (≥98 %), 2-phenylethylamine hydrochloride (≥98 %), cadaverine dihydrochloride (≥99 %), histamine dihydrochloride (≥99 %), tyramine hydrochloride (≥98 %), spermidine trihydrochloride (≥98 %), spermine tetrahydrochloride, agmatine sulfate salt (≥97 %), 1,7-diaminoheptane (Dha, 98 %), cross-linked polyvinylpolypyrrolidone (PVPP), and dansyl chloride (≥99 %) were obtained from Sigma (St. Louis, MO, USA). Sodium carbonate anhydrous (>99.5 %), sodium hydroxide (≥99 %), sodium hydrogen carbonate (>99 %), acetone (HPLC grade), acetonitrile (HPLC grade), and ammonia (25 %) were purchased from Merck (Darmstadt, Germany). From Fischer Scientific (Fair Lawn, NJ, USA), 1-N hydrochloric acid (HCl) solution was obtained. Ultra-pure water (18.2 MΩ cm) was produced using a Simplicity system from Millipore (Bedford, MA, USA). Filter paper no. 4 was obtained from Whatman (NJ, USA), and Millex polyvinylidene difluoride (PVDF) 13-mm syringe filters (0.45 μm) were purchased from Millipore. Raw materials, intermediate products, and beer samples were provided from two local microbreweries (brewery 1 and brewery 2) and a laboratory-scale brewery (brewery 3).
Standard and Derivatization Solutions
BA stock solutions were individually prepared in 0.1 N hydrochloric acid for a given concentration of 1 mg mL−1. Pooled standard solutions were prepared by aliquot dilution from stock solutions. All BA solutions kept refrigerated at 4 °C were stable at least for 20 days. 1,7-Diaminoheptane (internal standard) stock solution of 0.4 g L−1 was also prepared in 0.1 N HCl. Sodium carbonate-bicarbonate buffer, pH 10, was prepared weekly. Just prior to use, 10 mg mL−1 dansyl chloride solution (Dns-Cl) was prepared in acetone.
Sample Preparation and Derivatization
Raw materials: 3 g of malted barley (or oat), 2 g of hop, or 2 g of yeast was accurately weighed and transferred into a 100-mL Erlenmeyer flask; then, 100 μL (40 μg) of internal standard stock solution was added. Extraction was carried out with 20 mL of 0.1 N HCl in a Boeco (Staufen, Germany) OS-20 orbital shaker at 180 rpm for 30 min. To remove polyphenols and related substances, 5 mL of filtered extract (filter paper No. 4) was mixed and shaken with 250 mg of PVPP for 15 min at 180 rpm. After filtration (filter paper No. 4), 100 μL was used for derivatization. For intermediate products and beer samples, 50 mL was degassed in ultrasonic bath for 30 min at 20 °C. Fifty microliters (20 μg) of internal standard stock solution was added to 10 mL of degassed beer aliquot before the addition of 500 mg of PVPP. The mixture was shaken in a Boeco OS-20 orbital shaker for 15 min at 180 rpm and then filtered through filter paper No. 4. Into a 1.5-mL microtube, 100 μL of filtrate (or standard) was dansylated adding 400 μL of carbonate-bicarbonate buffer, pH 10.0, 300 μL of acetone, and 200 μL of Dns-Cl solution. The mixture was vortex-mixed during 30 s and then incubated for 60 min at 47 °C; afterward, the remaining dansyl chloride was removed (consumed) by the addition of 100 μL ammonia (25 % v/v). After 30-min reaction (protected from light), the sample was filtered through a 13-mm PVDF syringe filter (0.45 μm) and injected into a HPLC system.
Chromatography
BA analysis was performed using a Shimadzu (Kyoto, Japan) Prominence HPLC system consisted of the following: LC-20AT pump, DGU-20A5R degassing unit, CTO-20AC column oven, CBM-20A communication module, SPD-M20A diode array detector (DAD) coupled to Waters (Milford, MA, USA) 2475 multi-λ fluorescence detector, and Rheodyne (Cotati, CA, USA) 7725i manual injector with a 20-μL loop. Data were acquired and recorded by means of Shimadzu LabSolutions version 5.51 software Corporation and Waters Empower 2 software. Chromatography was carried on Agilent (Palo Alto, CA, USA) Zorbax Eclipse XDB-C18 (150 mm × 4.6 mm, 5 μm) column connected to Agilent Zorbax Eclipse XBD-CN (4.6 × 12.5 mm; 5 μm) guard column, both set at 40 °C, using a binary mobile phase composed of water (A) and acetonitrile (B) applying the following gradient program at a flow rate of 1.2 mL min−1: 0–5 min 30–40 % B, 5–15 min 40–60 % B, 15–19 min 60–60 B (isocratic step), 19–27 min 60–100 % B, 27–31 min 100–100 % B (isocratic step), and 31–32 min 100–30 % B, with 8 min of column conditioning. Detection was performed by UV absorption at 254 nm (His) and by fluorescence using 330 and 520 nm as excitation and emission wavelengths, respectively.
Statistical Analysis
Data were evaluated using descriptive statistics [means, standard deviation (SD) and relative standard deviation (RSD)]. Calibration equations were established using linear regression model relating biogenic amine concentrations (mg L−1) and the signal ratio from analyte and internal standard (1,7-diaminoheptane). Calibrations with and without matrix were compared using F test. Comparison of BA content was done using parametric tests, i.e., Student’s t or one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison test. All the above statistical analyses were carried out with a significance level (α) of 0.05 using GraphPad (San Diego, CA, USA) Prism 6.0 software. Central composite design (CCD) was prepared and analyzed by means of Statgraphics Centurion XV version 15.1.02 software (Rockville, MD, USA).
Results and Discussion
Sample Preparation
BA extraction from raw materials was carried out using 20 mL of 0.1 N HCl at ratios of 1:7 (w/v) for malted barley and oat and 1:10 (w/v) for hop and yeast. Lower ratios (higher sample amounts) diminished the extraction yields as well as produced chromatograms with higher backgrounds and interferences. The use of 0.1 N HCl as extraction solvent has been reported for different kinds of solid samples (Kalač and Křížek 2003; Onal et al. 2013), and its use is supported by its low toxicity and minor environmental impact compared to organic solvents. The cleanup step with PVPP was not relevant for malted barley, oat, and yeast, but its usefulness was clearly observed for hops, intermediate products, and beer samples, accomplishing lower-background chromatograms. Derivatization procedure was carried out following the method reported by Henríquez-Aedo et al. (2012) for BA determination in wines. This method showed repeatable results for all BAs (Agm, Phe, Put, Cad, His, Tyr, Spd, and Spm) in standard solution. This satisfactory performance was also observed in matrix (beer sample), except for Agm, which showed erratic and reduced yields. Derivatization conditions were unsuccessfully modified trying to accomplish adequate Agm yields, e.g., larger sample amounts (up to 350 μL), derivatization reagent volume (up to 500 μL), incubation temperature (up to 60 °C), and pH (up to 11). Higher pH values or other buffers were not evaluated because they reduce Spm and Spd derivatization yields (Pineda et al. 2012). Using specific conditions (350 μL of sample, 350 μL of Dns-Cl, and 300 μL of carbonate solution (8 % w/v)), it was possible to observe a weak Agm fluorescence signal at concentrations higher than 15 mg L−1. However, under these conditions, low levels (5 mg L−1) of Put, Cad, Spd, and Spm produced a saturation of fluorescence detector. Therefore, it was not possible to determine Agm at the concentrations commonly found in beers (<14 mg L−1) (Kalač and Křížek 2003). This unsuccessful reaction of Agm with dansyl chloride has been reported elsewhere (Anli et al. 2004; Burtin and Michael 1997; Ozdestan and Uren 2009). Nevertheless, other authors, e.g., Alberto et al. (2002) and Choi et al. (2012), have reported a successful dansylation of Agm standard solution (1.0 to 10.0 mg L−1), but without being detected in beer samples. Although other authors described additional steps to extract dansylated Agm using solvents or solid-phase extraction (Bencsik et al. 1998; Galgano et al. 2012; Proestos et al. 2008), these procedures were not assayed because they increase the analysis time and cost. Derivatization with o-phthalaldehyde was also experimentally tested achieving positive results for Agm but negative reaction for Spm and Spd (Ozdestan and Uren 2009). In view of these results, the use of dansyl chloride was preferred because it adequately reacts with two of three natural BA and it produces suitable and stable derivates.
Chromatographic Optimization
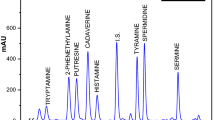
The development of a multi-matrix chromatographic method is difficult itself because it should be capable of separating and quantifying all analytes regardless of the matrix. Because the diversity of matrices used in brewing a wide range of interfering compounds can be found, further, some matrices such as hop and yeast are especially complexes. As described above, the matrices studied were as follows: malted barley, oat, hops, yeast, intermediate products, and beer. Even though the dansylation reaction has the advantage of providing suitable and stable signals, it is also a non-specific reaction interacting with different types of compounds, e.g., amino acids, carbonyls, and phenols, which made the method development difficult. Following the methodology proposed by Pineda et al. (2012), a mixture of water and acetonitrile was used as mobile phase with which different gradient programs, flow rates (0.4 to 1.4 mL min−1), and column temperatures (30 and 50 °C) were evaluated. After establishing the appropriate chromatographic conditions capable of separating all analytes (seven BAs and internal standard), different columns were assayed, i.e., Waters (Milford, MA, USA) YMC ODS C18 (150 × 4.6 mm; 5 μm), Waters Atlantis C18 (150 × 4.6 mm; 3 μm), Kromasil (Bohus, Suecia) 100–5 C18 (250 × 4.6 mm; 5 μm), Kromasil Eternity C18 (100 × 4.6 mm; 2.5 μm), and Agilent Zorbax Eclipse XDB-C18 (150 × 4.6 mm; 5 μm). With the last three columns, it was possible to achieve a clear separation as well as lower background chromatograms (data not shown). From them, Zorbax Eclipse XDB-C18 column was selected because it showed the best relation between the flow rate, backpressure, and time of analysis. Chromatographic conditions were optimized using a face-centered central composite design (CCD), which is a multivariate statistical model that shows a high efficiency with a reduced number of experiments (Ferreira et al. 2007). Following our previous approach (Galarce-Bustos et al. 2014), the evaluated responses were peak height and resolution, the first one with the purpose of enhancing detection limits and the second one to improve the analyte separation from matrix interference peaks. Considering these variables, three factors were studied: slope of acetonitrile at the initial part of the gradient program, expressed as the percentage of acetonitrile at 19 min (%ACN), column temperature (°C), and flow rate (mL min−1). In view of preliminary chromatographic assays, the following ranges were evaluated for each factor: %ACN in mobile phase (40–80 % v/v), column temperature (30–50 °C), and flow rate (0.8–1.4 mL min−1). Because chromatographic separation was done using a gradient program, the factor flow rate was assayed separately and set at 1.0 mL min−1 during optimization. Thus, the CCD experimental plan considered ten runs including two central points (Table 1). All runs were carried out in duplicate using a beer sample spiked with 10 mg L−1 of each analyte and 2 mg L−1 of internal standard. First, optimization was carried out individually for each response and BA. Optimization results showed that %ACN significantly influenced (p < 0.05) the response peak height of Phe and Cad and the resolution of Phe, Tyr, Spd, and Spm. Column temperature significantly affected (p < 0.05) the variable peak height of Phe and Put and the resolution of Phe. By means of individual optimum, a multiple response optimization was performed establishing the following optimal conditions for resolution (50.8 % of ACN and column temperature of 50 °C) and peak height (80.0 % ACN and 44.8 °C). Both optimal conditions provided maxima responses for all analytes and matrices except for Spm and Spd in hop matrix, which showed adjacent peaks (data not shown). To improve Spd and Spm resolution but maintaining adequate responses for all BAs, the following chromatographic conditions (also within the optimal range) were selected: column temperature of 40 °C and 60 % v/v of ACN at 19 min of gradient program. Flow rate was defined in 1.2 mL min−1 accomplishing an adequate equilibrium between resolution, time of analysis, and backpressure. With these optimal conditions, clear and well-resolved chromatograms were obtained for all matrices accomplishing a complete separation of all BAs and internal standard in less than 30 min (Fig. 1).

LC/FLD chromatograms of beer (a), yeast (b), malted barley (c), and hop (d) samples (lines) and the same samples spiked with 5 mg L−1 of each biogenic amine (dashed lines). Phe phenylethylamine, Put putrescine, Cad cadaverine, His histamine, Dah diaminoheptane (IS), Tyr tyramine, Spd spermidine, Spm spermine. Asterisks by-products
Validation
The analytical methodology was validated following the International Conference on Harmonization (ICH) recommendations (International Conference on Harmonisation (ICH) 2005). Calibration curves with and without matrix were compared in order to evaluate a possible matrix effect. Samples of beer, malted barley, hop, and yeast were spiked with different standard levels; the real peak heights and areas were calculated by subtracting the initial BA content of each matrix. In the case of yeast, a 20-fold dilution was necessary due to the high levels of Spd and Spm. After slope and intercept comparison (data not shown), statistical differences were only observed in Spd and Spm calibrations (p < 0.05). In order to develop a simple and fast method, calibrations were established without matrix, and Spd and Spm contents were corrected according to recovery evaluation (if necessary). Calibrations were established at five levels from 0.5 to 20.0 mg L−1. Each calibration level was evaluated using three independent samples, which were derivatized and injected in duplicated. A linear response was observed for all BAs over the examined concentration range with determination coefficients (R 2) from 0.9962 to 0.9999 (Table 2). His was also quantified by UV at 254 nm due to the low signal observed by fluorescence detection. Method precision was evaluated through repeatability and intermediate precision. Repeatability was assessed by repeated measurements (n = 6) of each spiked matrix (5 mg L−1), showing RSD values from 0.13 to 7.62 % (Table 2). Intermediate precision was evaluated analyzing in duplicate (n = 2) the same spiked samples (5 mg L−1) of beer, malted barley, hop, and yeast during three different days (n = 3) showing RSD values from 0.33 to 8.51 % (Table 2). Method accuracy was determined through recovery evaluation spiking each matrix with three different BA levels defined according to the calibration range and the reported values. Beer sample was spiked with 1.0, 5.0, and 10.0 mg L−1; malted barley with 6.7, 33.3, and 66.7 mg kg−1; and hops and yeast with 10.0, 33.3, and 100 mg kg−1. Each level was prepared daily and measured in triplicate during 3 days. Recovery values expressed as percentage showed the following intervals for the three levels: 73.97 ± 2.41 to 113.54 ± 1.53 % (first level), 75.75 ± 0.24 to 113.85 ± 2.47 % (second level), and 81.93 ± 3.71 to 114.94 ± 9.17 % (third level). RSD values were lower than 11.40 % (Table 2). Detection and quantification limits were calculated using signal to noise ratios (S/N) of 3 and 10, respectively. Considering an injection volume of 20 μL, the detection (LOD) and quantification (LOQ) limits ranged from 0.003 to 0.102 mg L−1 and from 0.009 to 0.340 mg L−1 for beer; 0.024 to 0.730 mg kg−1 and 0.080 to 2.530 mg kg−1 for malted barley, and 0.030 to 1.400 mg kg−1 and 0.100 to 4.60 mg kg−1for hop, respectively. For the case of yeast, LOD and LOQ were not calculated due to the high concentration of Spd and Spm, forcing to carry out a 20-fold dilution. Even though ICH describes the term specificity, due to general agreement and IUPAC recommendation, the preferred and promoted term is selectivity. The identification of BA peaks was carried out comparing retention time (t R), via standard addition method and fluorescence and UV absorption spectra, showing correlation coefficients (r) >98 %. Selectivity was evaluated measuring peak purity. Fluorescence spectra correlations (r) between peak start and peak apex and between peak apex and peak end showed, for both excitation and emission wavelengths, showed r > 0.99, for all BAs in all matrices. Additionally, peak purity was also determined by UV absorption purity index using DAD showing r > 0.99 for all BAs in all matrices. Thus, the optimized and validated multi-matrix chromatographic method is adequate for a reliable quantification of seven BAs during the entire brewing process (Fig. 1). Robustness was evaluated simultaneously with optimization using a response surface design. According to the statistical evaluation (ANOVA), both factors (%ACN and column temperature) showed a significant effect (p < 0.05) over the chromatographic response (peak height and resolution). Therefore, both parameters should be carefully controlled. Different solvent lots and precolumns from the same manufacturer did not show any relevant effect over chromatography.
Method Comparison
Comparison with other published methods is complicated because only few studies have reported the determination of BA during brewing process, and from them, only one analyzed all raw materials (Kalac et al. 1997). To the best of our knowledge, this is the first multi-matrix method reported for BA determination during the entire brewing process using dansyl chloride, which is one of the most suitable derivatization reagents for BA determination. In general, the reported methods showed similar validation results to those obtained with the proposed method. Kalac et al. (1997) analyzed all raw materials and beers using benzoyl chloride as derivatization reagents. Comparatively, this method required two extractive processes to evaluate five BAs and the derivatization process presents more steps than the proposed method including an extraction with diethyl ether. The linear range reported is similar to the one established here, but the first level is ten times superior (5–25 mg L−1). Determination coefficients (R 2>0.99) and precision (RSD ≤11.4 %) were in the same range, and LODs (0.3 mg L−1 for all the amines) were higher than those the observed with the proposed method. Cortacero-Ramirez et al. (2007) described a capillary zone electrophoresis method with detection via laser-induced fluorescence using fluorescein isothiocyanate (FITC) as derivatization reagent. BAs were determined during brewing process, but none of the raw materials (hops, yeast, malted barley, etc.) was analyzed. Therefore, it is not possible to compare extraction and chromatographic methods because the matrix complexity (interference levels) is completely different. Derivatization process with FITC is similar to the protocol applied for dansyl chloride, but the derivatization reaction requires 120 min, which is 2-fold the time reported in this study. Romero et al. (2003) reported the BA determination in some raw materials (malt and maize), intermediate products, and beer samples using dabsyl chloride (vis range). From validation data, it is possible to indicate that recovery (87.8 to 109.5 %) and precision (RSD ≤14.5) are similar to the values obtained in this study. From figures and tables, it is possible to infer that detection and quantification limits are in the same order of magnitude. Run time (ca. 46 min) is slightly higher than the one reported here. Hops and yeasts were not analyzed; therefore, as indicated before, it is not possible to carry out a suitable comparison. Other authors have also reported the evaluation of BA in raw materials and brewing, but due to the lack of validation data, it was not possible to carry out complete comparison (Halasz et al. 1999; Izquierdo-Pulido et al. 1994). Further, none of both methods analyzed all raw materials. Methods that only evaluated BA in beers were not included in this comparison because the interference level is completely different. BA analysis in beers is simpler, obtaining shorter analysis times and better detection and quantification limits (Dadakova et al. 2009).
Method Application
The optimized and validated method was applied to evaluate the dynamic of BA content in three different breweries (1–3). Brewery 1 has a production level of 1500 L per week, brewery 2, 500 L per week; and brewery 3, 20 L (laboratory scale). The latter was used as control (all hygienic conditions were highly controlled). All brewing processes were carried out using low fermentation (ale) producing red ale and stout beers. Samples were collected during the entire brewing process, i.e., raw materials, mashing, boiling, fermentation, maturation, and bottling. The analyses of raw materials included different types of malted barley, oat, hop, and yeast. BA concentration found in yeast can be used only as approximation due to different cell counts during the process (Kalac et al. 1997). Malted barleys showed a BA content from 155.63 to 214.56 mg kg−1, hops from 123.56 to 174.10 mg kg−1, and yeasts from 919.75 to 961.19 mg kg−1 (Table 3). In concordance with other reports, the most important BAs found in malted barley were Put, Spd, and Spm (Halasz et al. 1999; Izquierdo-Pulido et al. 1994; Romero et al. 2003). Hops showed similar profile but with higher Tyr levels, and in case of yeasts, the main BAs found were Spd and Spm (Table 3). The contribution of raw materials to BA content in the final product was calculated considering the quantity used and the volume produced. In agreement with Izquierdo-Pulido et al. (1994), the main contribution came from malted barley while hops and yeasts showed a minor influence due to the lower amounts added. Regarding the dynamic of BA content during brewing (Fig. 2), BA content in brewery 3 (control) decreased from raw materials to mashing (54.46 to 21.53 mg L−1). Individually, most BA showed the same behavior, which is concordant with already published reports (Halasz et al. 1999; Izquierdo-Pulido et al. 1994; Romero et al. 2003). Spd (19.45 to 3.88 mg L−1) and Spm (7.85 to 0.65 mg L−1) showed the highest decrease (Table 4). This decrease has been related to thermal decomposition (Romero et al. 2003) and biochemical transformations (Izquierdo-Pulido et al. 1994). Brewery 1 showed a similar trend decreasing from 53.39 to 18.79 mg L−1. Contrarily, brewery 2 showed a slight increase (53.83 to 55.18 mg L−1) due to increase of Cad (1.60 to 11.13 mg L−1) and Tyr (2.40 to 32.04 mg L−1) levels. Spd and Spm decreased in both breweries as observed also in control brewing (Table 4). The increase of Cad and Tyr concentrations is related to bacterial contamination during mashing. In brewery 3, the content of Put, Cad, and Tyr from boiling to bottling was constant, while Spd and Spm concentrations decreased at trace levels. His was not detected, and other bacterial BAs (principally Cad and Tyr) were found in low concentration. Brewery 1 showed a significant increase of Put (10.49 to 17.30 mg L−1), Cad (1.69 to 130.84 mg L−1), and His (0.69 to 4.23 mg L−1) from mashing to boiling; this increase could be associated with bacterial contamination (Halasz et al. 1999; Izquierdo-Pulido et al. 1994; Kalač and Křížek 2003; Romero et al. 2003). Put levels showed an important increase (2-fold) during fermentation remaining constant to bottling. The same His level achieved during boiling was observed in the following stages; therefore, no further bacterial contamination with aminogenic capacity was observed during maturation and bottling. Thus, using the proposed chromatographic method, it was possible to establish the dynamic of BA content in brewery 1 (Fig. 2), defining mashing, boiling, and fermentation as critical stages, and therefore, they should be better controlled. Brewery 2 showed a similar behavior than brewery 3 from boiling to bottling, which demonstrated adequate hygienic conditions (Fig. 2). However, during mashing, Cad and Tyr levels showed a significant increase, which is directly related to bacterial contamination. Accordingly, it was possible to determine that mashing was the critical stage for brewery 2 (Fig. 2).

Dynamic of total BA content during the entire brewing process
Conclusions
The present work reports for the first time (to the best of our knowledge) an optimized and validated chromatographic method to determine the dynamic of BA content during the entire brewing process using dansyl chloride, which is one of the most suitable and used derivatization reagents. Considering the matrix complexity, the method could be considered selective and reliable. The proposed method can be applied by any laboratory because it requires common reagents, solvents, and chromatographic columns. Furthermore, it can easily be transferred to HPLC/MS because it uses MS-compatible solvents. Applying this highly selective (multi-matrix) chromatographic method, it was possible to evaluate the dynamic of BA content during the entire brewing process (from raw materials to bottling) without any method adjustments. With the proposed method, it was possible to determine the critical steps (critical control points) of all breweries analyzed, defining the principal sources or stages of BA contribution/formation during productive process. Thus, breweries can carry out corrective actions, for example, improving the hygienic conditions and performing a careful control of fermentation process, especially the management of microbial starters (yeast in this case). The proposed method can also be used to control the yeasts used for brewing. Finally, this is the first time that the dynamic of BA content is determined in Chilean breweries.
References
Aflaki F, Ghoulipour V, Saemian N, Sheibani S (2014) Biogenic amine contents in non-alcoholic beers: screening and optimization of derivatization. Food Anal Method 7:713–720. doi:10.1007/s12161-013-9746-x
Alberto MR, Arena ME, de Nadra MCM (2002) A comparative survey of two analytical methods for identification and quantification of biogenic amines. Food Control 13:125–129
Almeida C, Fernandes JO, Cunha SC (2012) A novel dispersive liquid-liquid microextraction (DLLME) gas chromatography–mass spectrometry (GC-MS) method for the determination of eighteen biogenic amines in beer. Food Control 25:380–388. doi:10.1016/j.foodcont.2011.10.052
Alvarez MA, Moreno-Arribas MV (2014) The problem of biogenic amines in fermented foods and the use of potential biogenic amine-degrading microorganisms as a solution. Trends Food Sci Technol 39:146–155. doi:10.1016/j.tifs.2014.07.007
Anli RE, Bayram M (2009) Biogenic amines in wines. Food Rev Int 25:86–102. doi:10.1080/87559120802458552
Anli RE, Vural N, Yilmaz S, Vural YH (2004) The determination of biogenic amines in Turkish red wines. J Food Compos Anal 17:53–62
Anli RE, Vural N, Demiray S, Mert B (2006) Biogenic amine content of beer consumed in Turkey and influence of storage conditions on biogenic amine formation. J Inst Brew 112:267–274
Bencsik K, Kremmer T, Boldizsár M, Tamás J, Mák M, Páldi E (1998) High-performance liquid chromatographic determination and standardization of agmatine. J Chromatogr A 824:175–180. doi:10.1016/S0021-9673(97)00193-3
Bokulich NA, Bamforth CW (2013) The microbiology of malting and brewing. Microbiol Mol Biol Rev 77:157–172. doi:10.1128/mmbr.00060-12
Burtin D, Michael AJ (1997) Overexpression of arginine decarboxylase in transgenic plants. Biochem J 325(Pt 2):331–337
Camacho M, Torres G, Izquerdo P, Benitez B, Allara M, Céspedes E (2007) Determinacion de aminas biógenas en cervezas que se expenden en la ciudad de Maracaibo-Venezuela. Multiciencias 7:276–281
Choi S, Lee JK, Shukla S, Kim M (2012) Physiochemical properties and determination of biogenic amines in Korean microbrewery beer products. J Food Biochem 36:766–773. doi:10.1111/j.1745-4514.2012.00670.x
Cortacero-Ramirez S, Arraez-Roman D, Segura-Carretero A, Fernandez-Gutierrez A (2007) Determination of biogenic amines in beers and brewing-process samples by capillary electrophoresis coupled to laser-induced fluorescence detection. Food Chem 100:383–389. doi:10.1016/j.foodchem.2005.09.037
Dadakova E, Krizek M, Pelikanova T (2009) Determination of biogenic amines in foods using ultra-performance liquid chromatography (UPLC). Food Chem 116:365–370. doi:10.1016/j.foodchem.2009.02.018
Daniel D, dos Santos VB, Rajh Vidal DT, do Lago CL (2015) Determination of biogenic amines in beer and wine by capillary electrophoresis-tandem mass spectrometry. J Chromatogr A 1416:121–128. doi:10.1016/j.chroma.2015.08.065
Deetae P, Perello MC, de Revel G (2013) Occurrence of ochratoxin A and biogenic amines in Asian beers sold in French markets. J Inst Brew 119:57–63. doi:10.1002/jib.63
EFSA (2011) Scientific opinion on risk based control of biogenic amine formation in fermented foods. EFSA Panel on Biological Hazards (BIOHAZ). EFSA J 9:1–93
European Community (2011) BIAMFOOD: controlling biogenic amines in traditional food fermentations in Regional Europe. http://cordis.europa.eu/fetch?CALLER=FP7_PROJ_EN&ACTION=D&DOC=3&CAT=PROJ&QUERY=01309580ed03:40bc:04ab0338&RCN=88095. Accessed June 13, 2011
Fernandes JO, Judas IC, Oliveira MB, Ferreira I, Ferreira MA (2001) A GC-MS method for quantitation of histamine and other biogenic amines in beer. Chromatographia 53:S327–S331. doi:10.1007/bf02490351
Ferreira SLC et al (2007) Statistical designs and response surface techniques for the optimization of chromatographic systems. J Chromatogr A 1158:2–14
Galarce-Bustos O, Alvarado M, Vega M, Aranda M (2014) Occurrence of ochratoxin A in roasted and instant coffees in Chilean market. Food Control 46:102–107. doi:10.1016/j.foodcont.2014.05.014
Galgano F, Caruso M, Condelli N, Favati F (2012) Focused review: agmatine in fermented foods. Front Microbiol 3 doi:10.3389/fmicb.2012.00199
Gloria MBA, Izquierdo-Pulido M (1999) Levels and significance of biogenic amines in Brazilian beers. J Food Compos Anal 12:129–136
Halasz A, Barath A, Holzapfel WH (1999) The biogenic amine content of beer; the effect of barley, malting and brewing on amine concentration. Z Lebensm Unters Forsch 208:418–423. doi:10.1007/s002170050440
Henríquez-Aedo K, Vega M, Prieto-Rodríguez S, Aranda M (2012) Evaluation of biogenic amines content in Chilean reserve varietal wines. Food Chem Toxicol 50:2742–2750. doi:10.1016/j.fct.2012.05.034
International Conference on Harmonisation (ICH) (2005) ICH harmonised tripartite guideline validation of analytical procedures: text and methodology Q2 (R1)
Izquierdo-Pulido M, Marine-Font A, Vidal-Carou M (1994) Biogenic-amines formation during malting and brewing. J Food Sci 59:1104–1107. doi:10.1111/j.1365-2621.1994.tb08201.x
Kalač P, Křížek M (2003) A review of biogenic amines and polyamines in beer. J Inst Brew 109:123–128
Kalac P, Hlavata V, Krizek M (1997) Concentrations of five biogenic amines in Czech beers and factors affecting their formation. Food Chem 58:209–214. doi:10.1016/s0308-8146(96)00098-2
Loret S, Deloyer P, Dandrifosse G (2005) Levels of biogenic amines as a measure of the quality of the beer fermentation process: data from Belgian samples. Food Chem 89:519–525
Loukou Z, Zotou A (2003) Determination of biogenic amines as dansyl derivatives in alcoholic beverages by high-performance liquid chromatography with fluorimetric detection and characterization of the dansylated amines by liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J Chromatogr A 996:103–113
Onal A, Tekkeli SEK, Onal C (2013) A review of the liquid chromatographic methods for the determination of biogenic amines in foods. Food Chem 138:509–515. doi:10.1016/j.foodchem.2012.10.056
Ozdestan O, Uren A (2009) A method for benzoyl chloride derivatization of biogenic amines for high performance liquid chromatography. Talanta 78:1321–1326. doi:10.1016/j.talanta.2009.02.001
Pineda A, Carrasco J, Pena-Farfal C, Henriquez-Aedo K, Aranda M (2012) Preliminary evaluation of biogenic amines content in Chilean young varietal wines by HPLC. Food Control 23:251–257. doi:10.1016/j.foodcont.2011.07.025
Pospiskova K, Safarik I, Sebela M, Kuncova G (2013) Magnetic particles–based biosensor for biogenic amines using an optical oxygen sensor as a transducer. Microchim Acta 180:311–318. doi:10.1007/s00604-012-0932-0
Proestos C, Loukatos P, Komaitis M (2008) Determination of biogenic amines in wines by HPLC with precolumn dansylation and fluorimetric detection. Food Chem 106:1218–1224. doi:10.1016/j.foodchem.2007.06.048
Ramon-Marquez T, Medina-Castillo AL, Fernandez-Gutierrez A, Fernandez-Sanchez JF (2016) Novel optical sensing film based on a functional nonwoven nanofibre mat for an easy, fast and highly selective and sensitive detection of tryptamine in beer. Biosens Bioelectron 79:600–607. doi:10.1016/j.bios.2015.12.091
Romano A, Klebanowski H, La Guerche S, Beneduce L, Spano G, Murat M-L, Lucas P (2012) Determination of biogenic amines in wine by thin-layer chromatography/densitometry. Food Chem 135:1392–1396. doi:10.1016/j.foodchem.2012.06.022
Romero R, Bagur MG, Sanchez-Vinas M, Gazquez D (2003) The influence of the brewing process on the formation of biogenic amines in beers. Anal Bioanal Chem 376:162–167. doi:10.1007/s00216-003-1885-2
Spano G et al (2010) Biogenic amines in fermented foods. Eur J Clin Nutr 64:S95–S100. doi:10.1038/ejcn.2010.218
Acknowledgments
This work is part of Oscar Galarce thesis to obtain the degree of Magister in Pharmaceutical Sciences from the University of Concepcion. National Fund for Scientific and Technological Development (FONDECYT) project no. 1131080, Fund for Scientific and Technological Equipment (FONDEQUIP) project no. 130209, and the University of Concepcion financially supported this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by the National Fund for Scientific and Technological Development (FONDECYT) project No. 1131080 and by the Fund for Scientific and Technological Equipment (FONDEQUIP) project No. 130209.
Conflict of Interest
Oscar Galarce, Karem Henríquez-Aedo, Darlene Pettersen, Carlos Peña-Farfal, and Mario Aranda declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Rights and permissions
About this article

Cite this article
Galarce, O., Henríquez-Aedo, K., Peterssen, D. et al. A Selective Chromatographic Method to Determine the Dynamic of Biogenic Amines During Brewing Process. Food Anal. Methods 9, 3385–3395 (2016). https://doi.org/10.1007/s12161-016-0526-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-016-0526-2