
Abstract
In this article, ultrasonic-assisted cloud point extraction (UA-CPE) was used to the preconcentration of trace Ag(I) and Cd(II) in dried nut and vegetable samples. After ion association of Ag(I) and Cd(II) ions with 3,7-diamino-2,8-dimethyl-5-phenyl-phenaziniumchlorid (Safranin T) in the presence of excess potassium iodide (KI) at pH 6.0, the ternary complexes formed were quantitatively extracted into a micellar phase of poly(ethyleneglycol-mono-p-nonylphenylether) (PONPE 7.5) and then determined by flame atomic absorption spectrometry (FAAS). The matrix effects of possible some interfering ions on the method recovery and tolerance limits were also investigated. Under the optimized reagent conditions, the method was linearly dependent on the analyte concentrations in the range of 0.08–90 and 3–250 μg L−1 with limits of detection of 0.02 and 0.9 μg L−1 for Ag(I) and Cd(II), respectively. The reproducibility as the percent relative standard deviations (RSDs %) (0.5, 5.0, and 25 μg L−1 for Ag(I) and 15, 30, and 100 μg L−1 for Cd(II), n = 6) was lower than 3.6 % for both analytes. The accuracy of the method was controlled by analysis of two certified samples (SRM 1573a Tomato leaves and SRM 1643e Trace elements in waters) as well as recovery studies from spiked samples. It has been observed that the results obtained are in a good agreement with the certified values at confidence interval of 95 %. The method was successfully applied to determination of Ag(I) and Cd(II) in several dried nut and vegetable samples by FAAS after separation/preconcentration with UA-CPE.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Due to anthropogenic activities and rapidly growing industrialization in world, the foods (beverage, vegetables, nuts) and environmental (water, air, atmosphere) have been polluted by heavy metals and their compounds, and in most cases, highly toxic metals like silver (Ag(I)) and cadmium (Cd(II)) have been steadily increasing (Krizkova et al. 2009a). People can be exposed to these metals due to drinking water, ingestion of food, cigarette smoke, and cosmetic products. The result of studies shows that silver levels in most food samples can be found in the range of 10–100 μg kg−1, while the median daily intake of this metal has been estimated at a level of 7.1 μg kg−1 (Garbos and Swiecicka 2013). The toxicity of Ag(I) ions for human health is probably caused by their very good affinity for nucleic acids and proteins. The binding into an active site of the enzyme leads to the expressive inhibition of enzyme activity (Krizkova et al. 2009b). The toxicology of Cd(II) has destructive effects like liver, lung, kidney, and heart problems on human (Olmedo et al. 2013). According to a report of the World Health Organization (WHO), weekly intake of Cd(II) should be 7.0 μg kg−1 body weight for all human groups (Kaličanin 2009). Although these metals are usually present in foods and vegetables at trace concentrations, long-term exposure can have negative human health impacts. Therefore, the accurate, sensitive, and selective determinations with a suitable method of Ag(I) and Cd(II) in the real-time samples are an important topic for analytical chemists.
Because Ag(I) and Cd(II) are present in trace amounts in foods and vegetables, the preconcentrations of these metals in the sample are needed to the reduction of matrix effect and increasing of their concentrations. This study was considered and used for separation and preconcentration of metal ions from the sample matrix using the ultrasonic-assisted cloud point extraction (UA-CPE). When compared with preconcentration procedures like liquid-liquid extraction, solid-phase extraction, and precipitation, the UA-CPE has the advantages like low cost, safety, high preconcentration factor, easy and fast operation, and no need for a large amount of organic solvents (Altunay et al. 2015). The preconcentration procedure is based on the heating of a non-ionic surfactant-containing solution under ultrasonic power. When the solution in ultrasonic bath is heated above a certain point (cloud point temperature), surfactant monomers form micelles; the solution becomes turbid and separates in two liquid phases by centrifugation. The small volume phase obtained after centrifugation is called surfactant-rich phase which includes micelles and analyte ions. The surfactant-rich phase containing the analyte is suitable to determine the amount of analytes with many analytical instruments such as flame atomic absorption spectrometry (FAAS) (Naeemullah et al. 2016); electrothermal atomic absorption spectrometry (ETAAS, which is also known as graphite furnace atomic absorption spectrometry (GFAAS)) (López-García et al. 2014; 2015), which involves introducing a small amount of sample and then resistively heating a tube of graphite, which becomes the atomization cell; and inductively coupled plasma optical emission spectrometry (ICP-OES) (Silva et al. 2015).
Determination of trace levels of Ag(I) and Cd(II) in water, foods, and vegetables is possible using several analytical techniques such as potentiometric sensors in complex mixtures (Shirmardi-Dezaki et al. 2012); FAAS in various food samples (Mashhadizadeh et al. 2014); inductively coupled plasma optical emission spectroscopy (ICP-OES) in mushrooms (Chudzyński et al. 2013); reversed-phase high-performance liquid chromatography (RP-HPLC) with UV-vis detection in water (Hu et al. 2003); GFAAS in biological, water, and soil samples (Yang et al. 2009); ETAAS in environmental and rice samples (Rahman et al. 2004; Silva et al. 2011); and inductively coupled plasma mass spectrometry (ICP-MS) in foods (Nardi et al. 2009). ETAAS (or GFAAS), ICP-MS, and ICP-AES are the most sensitive techniques for the determination of trace level of metals, but these techniques, especially ETAAS (or GFAAS), are expensive (when considered cost per analysis of both few elements and many elements in high volume), have poor short-term precision (0.5–5 % for 5–10 min) and long-term precision (1.0–10 % for 1–2 h), are time-consuming due to long furnace program, and need expert’s user in his/her area. However, ICP-based systems (ICP-OES or ICP-MS) require clean room conditions to operate at the ultra-trace levels, high-volume gas installations, and even highly high-purity reagents. Also, they require to use internal standard and costly method of isotope dilutions in order to improve poor precision. Unlike these techniques, the FAAS is a very attractive element-selective detection technique for determination of metal and metalloids in environmental, food, and biological samples due to low cost, ease of use and method development, low matrix effect, precision, accuracy, and high sample throughput (10–15 s per element).
In this study, a new UA-CPE approach was developed for the separation/preconcentration of Ag(I) and Cd(II) ions from the dried nut and vegetable samples prior to determination by FAAS. The method is based on the extraction of the Ag(I) and Cd(II) ions as hydrophobic ternary complexes from aqueous solution using poly(ethyleneglycol-mono-p-nonylphenylether) (PONPE 7.5) in the presence of 3,7-diamino-2,8-dimethyl-5-phenyl-phenaziniumchlorid (Safranin T) and excess potassium iodide (KI) at pH 6.0 and then their detections by FAAS after dilution of the surfactant-rich phase with ethanol. The variables affecting complex formation and extraction efficiency were optimized in detail. Under the optimized conditions, the method developed was successfully applied to analysis of the selected sample matrices and two certified samples.
Experimental
Instrumentation
Determinations of Ag(I) and Cd(II) in dried nut samples were carried out using an AAS-6300 (Shimadzu, Kyoto, Japan) flame AAS equipped with deuterium lamp, which is used as background correction, hollow cathode lamps, which are used as the radiation source for each element, and an air-acetylene flame atomizer. The instrumental operating conditions of the AAS spectrometer for Ag(I) were wavelength, 328.1 nm; lamp current, 10 mA; spectral bandwidth, 0.7 nm; burner height, 7 mm; and acetylene and air flow rates, 2.2 and 15.0 L min−1, respectively, while they were wavelength, 228.8 nm; lamp current, 8 mA; spectral bandwidth, 0.7 nm; burner height, 7 mm; and acetylene and air flow rates, 1.8 and 15 L min−1 for Cd(II), respectively. The nebulizer flow rate and burner height were adjusted in order to obtain the maximum absorbance signal by aspirating a solution containing the analytes in ethanol containing 0.1 mol L−1 HNO3. A Selecta pH meter (Barcelona, Spain) 2005 Model with glass electrode was employed for measuring pH values in the aqueous phase. An ultrasonic cleaner operating in the range of 0–80 °C with a frequency of 40 kHz at ultrasonic power of 300 W (UCP-10 model, Seoul, Korea) was used for preconcentration experiments and digestion of the samples, and a centrifuge (Hettich Universal, England) was utilized to accelerate the phase separation process. A vortex mixer with variable speed control in 0–3200 rpm (with a frequency of 50 Hz at power of 12 W) (VM-96B model, Seoul, Korea) was used to fasten the extraction process and minimize the reagent consumption during experimental procedures.
Reagents
Ultra-pure water (18.2 MΩ cm) obtained from a Labconco (Kansas City, USA) water purification system was used in this study. All chemicals were also guarantee reagent and purchased from Sigma St. Louis, MO (USA) unless otherwise stated in this study. The Ag(I) and Cd(II) stock standard solutions (1000 mg L−1) were prepared by dissolving in water appropriate amounts of AgNO3 and CdCl2, respectively. The working solutions were obtained by stepwise dilutions from stock solutions of Ag(I) and Cd(II). For the preparation of pH 6.0 phosphate buffer solution, 100 mL of 0.1 mol L−1 KH2PO4 and 11.2 mL of 0.1 mol L−1 NaOH solutions were mixed, completing to a final volume of 200 mL. A 1.0 × 10−3 mol L−1 of Safranin T solution was also prepared daily by dissolving with water. A 2.5 % (v/v) PONPE 7.5 solution was prepared by diluting 2.5 mL of compound in 20 mL ethanol and completing the volume to 100 mL with the water. A 0.02 mol L−1 solution of KI was prepared by dissolving a suitable amount of solid reagent in a calibrated flask of 250 mL with the water and was kept in a cool dark place when not in use. The laboratory glassware and PET bottles were soaked in a 5.0 % (v/v) HNO3 solution for 24 h and rinsed with the water at least five times prior to use.
The UA-CPE Procedure
A typical UA-CPE required the following steps: an aliquot of 50 mL of a solution containing Ag(I) 0 (0.08–90 μg L−1) and Cd(II) (3–250 μg L−1), 0.3 % (v/v) of PONPE 7.5, 0.005 mol L−1 KI, 0.1 × 10−4 mol L−1 of Safranin T, and 1.2 % (w/v) of NaCl were adjusted to pH 6.0 with phosphate buffer solution and then completed to 50 mL by the water. The mixture was shaken using a vortex agitator at 3000 rpm for 5 min and left to stand in an ultrasonic effect at 40 °C for 10 min. Separation of the phases was then achieved by centrifugation at 4000 rpm for 10 min. After cooling in a refrigerator for 5 min, the surfactant-rich phase became viscous. In this way, the bulk aqueous phase was easily decanted. To reduce the viscosity of the surfactant-rich phase and facilitate introduction to the nebulizer of FAAS, 1.0 mL of ethanol was added to the surfactant-rich phase using a vortex agitator. Finally, the Ag(I) and Cd(II) contents of the selected samples were readily evaluated by using either directly the calibration curve procedure obtained by FAAS or the standard addition curve procedure in order to control the matrix effect around detection limits when necessary.
Sample Collection
The determinations of Ag(I) and Cd(II) by FAAS were evaluated by analysis of samples such as dried nut (hazelnut, walnut, sauce peanut, peanut, roasted chickpea, cashew, sauce corn, and almond) and vegetable samples (spinach, lentils, leeks, cabbage, and parsley). All the samples were supplied from a local supermarket in Sivas, Turkey.
Ultrasonic-Assisted Sample Preparation
Sample Preparation for Dried Nuts
All of the selected dried nut samples (5.0 g) were homogenized through a stainless blender. The homogenized samples were transferred to a 100-mL flask. To simplify the process, minimize reagent volume consumption, temperature, and digestion time according to the type, nature, and composition of sample matrix, a slight modification of procedure suggested by Horwitz (2000) for digestion of vegetable samples, was adopted in this study. Due to relatively low oxidation potential, for efficient and complete destruction of materials with complex organic-rich matrices such as carbohydrates and fats, the combination of HNO3 with stronger oxidant, HClO4, was considered. With this purpose, 10 mL of 3.0 mol L−1 of HNO3 and 5.0 mL of 2.5 mol L−1 HClO4 were added to the sample and were then completed to 100 mL with the water. The mixtures were thoroughly sonicated and extracted under ultrasonic effect (with ultrasound frequency of 40 kHz at 300 W) at 80 °C for about 15 min until a clear solution is obtained. After cooling to room temperature, 4 mL of 3.0 mol L−1 of HNO3 and 2.0 mL of 2.5 mol L−1 HClO4 were added for complete digestion of the sample and again extracted under ultrasonic effect at 80 °C for 15 min. Then, the resulting clear sample solution was filtered using a membrane filter of 0.45 μm. The pH of the sample was adjusted to 7.0 using NaOH (0.5 mol L−1) and was completed to 50 mL with the water.
Sample Preparation for Vegetables
Firstly, the vegetables (10 g) were washed thoroughly with ultra-pure water, dried at 90 °C, and ground to pass a 200-mesh sieve. Five grams of the homogenized samples was transferred to a 100-mL flask. For digestion, 5.0 mL of 3.0 mol L−1 of HNO3 and 5.0 mL of 1.0 mol L−1 HClO4 were added to the sample and were then completed to 100 mL with the water. The mixtures were thoroughly sonicated and extracted under ultrasonic effect (300 W, 40 kHz) at 70 °C for about 15 min. When necessary, same procedure has been repeated until a clear solution is obtained. After cooling to room temperature, the resulting mixtures were filtered using a membrane filter of 0.45 μm. The pH of the samples was adjusted to 7.0 using NaOH (0.5 mol L−1) and was completed to 50 mL with the water. The above-mentioned sample preparation procedure was also applied to extraction of Ag(I) and Cd(II) contents of two certified samples (SRM 1573a Tomato leaves and SRM 1643e Trace elements in waters). The blank of the reagents was carried out following the same procedure without dried nut and vegetable samples. All measurements and processing were performed at least in five times.
Results and Discussion
General Aspects Related to Method Development
From prior studies, it was observed that Ag(I) and Cd(II) ions gave stable anionic complexes, AgI2 − and CdI3 −, with excess iodide (Hosseini and Hamid 2005; Gürkan and Altunay 2013), so as to give a maximum signal for both species in presence of Safranin T before preconcentration at pH 6.0. In fact, the formed anionic stable iodide complexes react with monomer forms of Safranin T to form the hydrophobic ion-pairing complexes depending on iodide concentration that can easily be extracted into surfactant-rich phase of Ponpe 7.5. Safranin T with pKa values of 3.0 and 8.5 is a cationic phenazine group dye (Guha and Mittal 1992, 1997; Borsarelli et al. 2002). At pH 6.0, it is clear that Safranin T is predominantly in the form of SFH+ when its acid dissociation equilibriums are considered. Also, it is clearly implied that cationic SFH+ monomer forms aggregate with negatively charged hydrophobic anionic surfactants such as sodium dodecyl sulfate (SDS), sodium dodecyl benzene sulfonate (SDBS), and Triton X-100 in the premicellar/micellar region, below and above the critical micellar concentration (CMC), and based on this aggregate formation with an adsorption constant of 1.80 × 105 on the micellar surface, it is used efficiently in the determination of anionic surfactant in the literature (Borsarelli et al. 2002; Gao et al. 2002; Asok and Jana 2004). In light of all these pieces of information, it can be concluded that the proposed method is based on micellar-enhanced atomic spectrometric monitoring of the ion-pairing complexes produced by efficient binding of SFH+ to anionic Ag(I)- and Cd(II)-iodide complexes, which is linearly related to concentration of each analyte in a wide linear range. The present properties of these ion-pairing complexes prompted us to study ion-pairing complexes with the aim of developing a new analytical method that could be useful in determination of Ag(I) and Cd(II) ions in real-time samples. In this sense, for further studies, the different variables affecting complex formation and extraction efficiency were optimized in order to achieve maximum sensitivity. Finally, the usefulness of the method was successfully indicated by analysis of the Cd(II) and Ag(I) in selected sample matrices by FAAS. The accuracy was also assessed by the analysis of two certified samples and recoveries of spiked samples after pretreatments.
Study of the UA-CPE Conditions
A comprehensive working has been carried out to optimize the parameters for the simultaneous preconcentration of Ag(I) and Cd(II) ions. The optimized experimental conditions for determination of low levels of Ag(I) and Cd(II) by FAAS are shown in Table 1.
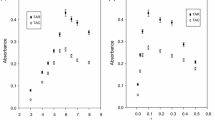
The pH is the most important factor to obtain hydrophobic complex between the ligand and metal ions. The effect of pH on UA-CPE efficiency has been carried out by changing the pH of aqueous solution of sample with different buffer systems such as citric acid/NaOH (in the range of 2.5–7.0), a mixture of citric acid-phosphoric acid-boric acid (in the range of 2.5–8.0), and phosphate buffer (in the range of 4.0–7.0). The phosphate buffer for fixed concentrations of Ag(I) and Cd(II) was selected to give the maximum absorbance. The results of the study for phosphate buffer are shown in Fig. 1a. It was found that, at pH of 6.0, the best recovery for each ion was achieved using phosphate buffer. The decrease in extraction efficiency at higher pH values than 6.0 may be due to hydrolysis of Cd(II) and Ag(I) ions with acid hydrolysis constant of 10.15 and 11.70, respectively (Foti et al. 2011), so as to give further Cd(OH)2, Cd(OH)3 −, and Cd(OH)4 2− species (or Ag(OH), Ag(OH)2 −, Ag(OH)3 2−, and Ag(OH)4 3−) as metal species with four coordinations. Thus, phosphate buffer was added to the sample solutions to maintain at pH 6.0.

a–d The individual optimizations of the analytical variables affecting the percent recoveries of Ag(I) and Cd(II) ions
Extraction of Ag(I) and Cd(II) ions at pH 6.0 requires a chelating agent to form an appropriate complex with these ions. Thus, KI was used as the primary soft binding ligand for Ag(I) and Cd(II) ions as soft metals in this study. The effect of concentration of KI on the recovery of Ag(I) and Cd(II) ions was studied in range from 0.002 to 0.01 mol L−1, and the results are shown in Fig. 1b. It was observed that the recoveries of Ag(I) and Cd(II) increased as the concentration changes from 0.002 to 0.01 mol L−1 and then remained constant. The recovery for analyte ions linearly decreased at the lower concentrations than 0.005 mol L−1. The cause of this decrease in recovery may be ion-association of iodide ions as a hydrophobic ionic species (owing to a large ion radius) with cationic Safranin T in absence of analyte ions, so as to cause an increase in blank signal. To achieve maximum recovery, thus, a KI concentration of 0.005 mol L−1 was chosen as optimal.
The UA-CPE is used to separate and preconcentrate metal ions from the sample matrix after the formation of water-insoluble complexes between the metal ions and the chelating ligand. Thus, Safranin T was selected as an ion-pairing reagent efficiently to bind a stable anionic Ag(I)- and Cd(II)-iodide complexes, AgI2 − and CdI3 − (or CdI4 2−, AgI3 2−, and AgI4 3− as depending on iodide concentration), at pH 6.0 in this study. The ion-pairing’s concentration was evaluated in the range of (0.05–0.25) × 10−4 mol L−1 for its effect on the recovery of metal ions. The results are represented in Fig. 1c. The maximum recovery for each ion was obtained at the concentration of 0.1 × 10−4 mol L−1 of the Safranin T. At higher concentrations than this value, the recovery and micelle formation for each metal was found to be decreased due to aggregation of dye and to be used more ethanol as the solvent of ion-association complex. To achieve maximum recovery, thus, a Safranin T concentration of 0.1 × 10−4 mol L−1 was chosen as optimal.
The non-ionic surfactant concentration as extracting agent is one of the main parameters affecting the phase volume ratio for a successful UA-CPE. In this study, the PONPE 7.5 with critical micelle concentration (CMC) of 0.085 mmol L−1 was selected as the non-ionic surfactant because of its commercial availability in a high purified homogeneous form and low toxicological properties and cost. The UA-CPA efficiencies of Ag(I) and Cd(II) using the PONPE 7.5 concentration were evaluated within the range of 0.02–1.0 % (v/v) in Fig. 1d. The highest recovery for each ion was obtained using 0.3 % (v/v) PONPE 7.5. Thus, a PONPE 7.5 concentration of 0.3 % (v/v) was chosen as optimal in this study.
The addition of an electrolyte to aqueous micellar solutions of the PONPE 7.5 usually reduces the cloud point due to the salting-out effect. Moreover, the hydrophobic ion-association complexes obtained for Ag(I) and Cd(II) may become less soluble in the aqueous solution at higher electrolyte concentration and thus contribute to higher extraction yield (Sosa-Ferrera et al. 2004). Thus, the effects of the different electrolyte solutions (NaCl, K2SO4, CaCl2, and NaF) on the recovery of Ag(I) and Cd(II) ions were studied; the best recovery for each ion was obtained using 1.2 % (w/v) NaCl. To achieve maximum recovery, thus, a NaCl concentration of 1.2 % (w/v) was chosen as optimal.
The equilibration temperature and incubation time of the experimental conditions can affect the ion-association reactions and micelle formation. The mechanism of phase separation for non-ionic surfactants is due to an increase in the micellar aggregation number when temperature is increased and can be caused by a change in micellar interactions (Ghaedi et al. 2009). This phenomenon can be explained on the basis of the dehydration process that occurs in the external layer of the micelles of non-ionic surfactants depending on the temperature increase. To achieve easy phase separation and preconcentration for Ag(I) and Cd(I), optimal equilibration temperature and incubation time were investigated using ultrasonic power (300 W, 40 kHz) in the range of 30–70 °C and 2–30 min, respectively. The results obtained from the optimization experiments for each analyte showed that equilibration temperature of 40 °C and incubation time of 10 min are adequate for the best extraction recovery and quantitative complexation.
To determine the optimal vortex time and rates, the centrifuge tubes were placed in the vortex mixing before ultrasonic bath. The effect of vortex time and rates on recovery of Ag(I) and Cd(II) was investigated in the time range of 0–10 min and 1000–3500 rpm, respectively. A vortex time of 5 min and vortex rate of 3000 rpm were found to be adequate to the best recovery and quantitative complexation. It has been seen that the fine droplets formed during the preconcentration procedure are capable of extracting the target analytes faster toward equilibrium because of the shorter diffusion distance and larger specific surface area.
The surfactant-rich phase obtained after the preconcentration has both high viscosity and rather small volume. For the determination of Ag(I) and Cd(II) ions in the surfactant-rich phase by FAAS, it is not sufficient to small volume. In addition, it causes clogging at introduction into the nebulizer of FAAS. For these reasons, it must be diluted with the appropriate diluents. As a result of studies carried out by using different diluents like methanol, ethanol, acetonitrile, acidic methanol, and acidic ethanol, the most suitable diluent was obtained using 1.0 mL of ethanol.
The Analytical Figures of Merit of the Method
The analytical figures of merit of UA-CPE/FAAS method were determined for Ag(I) and Cd(II) under the optimized reagent conditions. For this purpose, the important parameters such as the regression equations, linear working range, limits of detection and quantification, precision, sensitivity improvement, and preconcentration factors were determined to evaluate the analytical performances of the method for each analyte. The limits of detection (LODs) and quantification (LOQs) defined as 3σ blank/m and 10σ blank/m (where σ blank is the standard deviation of ten replicate measurements of the blank and m is the slope of the calibration graphs) were found to be 0.02 and 0.08 μg L−1 for Ag(I) and 0.9 and 2.8 μg L−1 for Cd(II), respectively. The sensitivity improvement factors for Ag(I) and Cd(II) were found to be 103 and 65, respectively. The reproducibility (as the percent relative standard deviations (RSDs %)) resulted from the analysis of six replicates of the 50-mL solutions containing 0.5, 5.0, and 25 μg L−1 for Ag(I) and 15, 30, and 100 μg L−1 for Cd(II) was in the range of 2.2–3.6 % and in the range of 1.8–3.2 %, respectively. The other results obtained are given in Table 2.
Matrix Effects
Because of the hollow cathode lamp used as the radiation source for each element in FAAS, any spectral interference in determining step is not observed. The matrix effects usually occur in the preconcentration step. The effect of possible matrix ions in the selected samples, in terms of some transition metals and anions, was also investigated to obtain the best recovery of Ag(I) and Cd(II). This study was conducted in the following manner. The interference effect of ions was investigated by adding known concentrations of each ion in a solution containing analytes and then determining the latter. Tolerance limit was defined as the highest amount of interference ions that produced an error not exceeding ±5.0 % in the preconcentration of investigated analyte ions by UA-CPE procedures and determination by FAAS. As can be seen from Table 3, it is clear that the method is fairly selective in terms of major species present in real-time samples. The possible interference effects of cations or anions can be avoided by the addition of suitable masking agents such as EDTA and reducing agents.
Accuracy, Precision, and Analytical Applications of the Method
The accuracy of the method was validated using two certified samples such as SRM 1573a Tomato leaves and SRM 1643e Trace elements in waters, in which their contents of Ag(I) and Cd(II) were determined by FAAS after preconcentration by UA-CPE from the digested samples under the ultrasonic effect. The results obtained are shown in Table 4. The results for Ag(I) and Cd(II) were statistically in good agreement with the certified values. For five replicate measurements, comparison of the means gave the calculated t values < the critical t values (2.78) for Ag(I) and Cd(II). Thus, the null hypothesis was accepted at 95 % confidence level in terms of the means found for each ion. In addition, the accuracy of the method was also evaluated by analysis of the dried nut and vegetable samples spiked at known concentration levels. The reliability was also checked by recovery experiments from spiked samples. The recovery rates of spiked samples are satisfactorily reasonable, which indicates the capability of the system in the determination of Ag(I) and Cd(II) in selected samples.
The intra-day and inter-day studies have been performed to evaluate the precision of the proposed method. In this study, pistachios and lettuce samples were evaluated under the optimal conditions. The measurements for selected samples were taken six times in 1 day for the intra-day precision and once a day on five consecutive days for the inter-day precision, respectively. The results are presented in Table 5. The intra- and inter-day precisions for pistachios expressed as RSDs % are less than 3.1 % and less than 4.0 %, respectively. The intra- and inter-day precisions for lettuce are less than 2.6 % and less than 3.3 %, respectively.
In order to demonstrate the applicability of the method, it was used for determination of trace amounts of Ag(I) and Cd(II) in dried nuts and vegetables by means of a standard addition method. The results summarized in Table 6 suggest that good recoveries for Ag(I) and Cd(II) spiked to the samples are achieved. The concentration of Cd(II) exceeding tolerable limit (7.0 μg kg−1) in vegetables and foodstuffs causes serious health problems such as kidney failure. The Cd(II) contents found varied from 19.7 to 69.4 μg kg−1 with RSD value ranging from 3.0 to 5.4 % for the selected vegetables while they varied from 3.7 to 12.1 μg kg−1 with RSD value ranging from 1.9 to 3.2 % for foodstuffs. Due to its bacteriostatic property, Ag(I) is often used in the processing of foods, drugs, and beverages. The concentration of Ag(I) exceeding tolerable limit (7.1 μg kg−1) in vegetables and foodstuffs can cause toxic effects on soft tissues although it is available at low concentrations in waters due to its low mobility. The Ag(I) contents found varied from 1.2 to 9.3 μg kg−1 with RSD value ranging from 3.7 to 5.0 % for the selected vegetables while they varied from 0.2 to 0.8 μg kg−1 with RSD value ranging from 1.9 to 3.7 % for foodstuffs. The concentrations found for Ag(I) except for cabbage and parsley with values of 7.7 and 9.3 μg kg−1, respectively, were within the permissible limits and safe in consumption point of view. However, the results show that the concentration of Cd(II) is approximately 10-fold higher than tolerable limit value of 7.0 μg kg−1 in all the selected vegetables including roasted chick, walnut, sauce corn, and almond ranging from 8.1 to 12.1 μg kg−1. To minimize the potential health risks resulted from contamination of heavy metals in growing vegetables in areas close to industry, it is clear that serious measures must be taken.
In terms of analytical performance as linear working range, preconcentration factor, reproducibility, LOD, and good precision especially at low concentrations, the performances of the proposed preconcentration method in Table 7 were compared with those of other methods present in literature. When examined Table 7, the LOD and reproducibility are better or comparable to some of the previously reported methods, and sensitivity improvement factors of 103 and 65 for Ag(I) and Cd(II), respectively, with preconcentration factor of 50are significantly higher than those of other methods. These characteristics are of great interest for the routine laboratories in the determination of trace amounts of Ag(I) and Cd(II). In terms of these analytical properties, the proposed method can be alternative to sensitive, but expensive detection techniques requiring expert user in his/her area such as ETAAS, ICP-OES, and ICP-MS. In this sense, the method presents simultaneous extraction, preconcentration, ease of use, short sample throughput, and low cost as the main advantages, particularly if more sophisticated techniques such as ETAAS or ICP-MS are not available.
Conclusions
In the present study, the UA-CPE was efficiently used for separation and preconcentration of trace amounts of Ag(I) and Cd(II) from dried nut and vegetable matrices. Also, the method was successfully applied to the determination of Ag(I) and Cd(II) by FAAS in the samples with relatively high sensitivity improvement and preconcentration factors and good reproducibility. Safranin T as an ion-pairing reagent was, for the first time, used to determine trace amounts of Ag(I) and Cd(II) in this study. The method has the following advantages: simplicity, ease of method development and use, low cost, simple apparatus, less organic waste, and short extraction/analysis times. In addition, the results of the study for Ag(I) and Cd(II) showed good reproducibility in analytical data, with RSD value of lower than 3.6 % on five repeated measurements, and satisfactory results of trace analysis of Ag(I) and Cd(II) in dried nut and vegetable samples were obtained.
References
Altunay N, Gürkan R, Orhan U (2015) A new ultrasonic-assisted cloud-point-extraction procedure for pre-concentration and determination of ultra-trace levels of copper in selected beverages and foods by flame atomic absorption spectrometry. Food Addit Contam A 32(9):1475–1487
Arvand M, Pourhabib A, Afshari A, Bagherinia M, Ghodsi N, Shemshadi R (2008) Determination of cadmium and zinc in water samples by flame atomic absorption spectrometry after cloud-point extraction. J Anal Chem 63(8):954–959
Asok K, Jana SR (2004) Studies on the molecular interaction of phenazine dyes with Triton X-100. Spectrochim Acta A 60:2093–2097
Biparva P, Hadjmohammadi RM (2011) Selective separation/preconcentration of silver ion in water by multiwalled carbon nanotubes microcolumn as a sorbent. Clean-Soil Air Water 3(12):1081–1086
Borsarelli CD, Bertolotti SG, Previtali CM (2002) Thermodynamic changes in the photoinduced proton-transfer reaction of triplet state of Safranine T. Photochem Photobiol Sci 1:574–580
Bosch-Ojeda C, Sánchez-Rojas F, Cano-Pavón JM (2010) Preconcentration of cadmium in environmental samples by cloud point extraction and determination by FAAS. Am J Anal Chem 1:127–134
Chamsaz M, Atarodi A, Eftekhari M, Asadpour S, Adibi M (2013) Vortex-assisted ionic liquid microextraction coupled to flame atomic absorption spectrometry for determination of trace levels of cadmium in real samples. J Adv Res 4:35–41
Chudzyński K, Jarzyńska G, Falandysz J (2013) Cadmium, lead and some other trace elements in Larch Bolete mushrooms (Suillus grevillei) (Klotzsch) Sing., collected from the same site over two years. Food Addit Contam B 6(4):249–253
Dadfarnia S, Shabani AMH, Kazemi E, Ahmad S, Khormizi H, Tammadon F (2015) Synthesis of nano-pore size Ag(I)-imprinted polymer for the extraction and preconcentration of silver ions followed by its determination with flame atomic absorption spectrometry and spectrophotometry using localized surface plasmon resonance peak of silver nanoparticles. J Braz Chem Soc 26:1180–1190
Filik H, Dondurmacıoglu F, Apak R (2008) Micelle mediated extraction of cadmium from water and tobacco samples with glyoxal-bis (2-hydroxyanil) and determination by electrothermal atomic absorption spectrometry. Int J Environ Anal Chem 88:637–648
Foroozan H, Masoumeh T, Zahra A, Hamed K, Samimi R (2014) Cloud point extraction and spectrophotometric determination of cadmium in some vegetables. Iran J Anal Chem 1:44–49
Foti CG, Lando FJ, Sammartano S (2011) Experimental study and modeling of inorganic Cd2+ speciation in natural waters. Environ Chem 8(3):320–323
Gao HW, Ye QS, Liu WG (2002) Langmuir aggregation of Nile blue and Safranine T on sodium dodecylbenzenesulfonate surface and its application to quantitative determination of anionic detergent. Anal Sci 18:455–459
Garbos S, Swiecicka D (2013) Human exposure to silver released from silver-modified activated carbon applied in the new type of jug filter systems. Rocz Panstw Zakl Hig 64(1):31–36
Ghaedi M, Shokrollahi A, Niknam K, Niknam E, Najibi A, Soylak M (2009) Cloud point extraction and flame atomic absorption spectrometric determination of cadmium (II), lead (II), palladium(II) and silver(I) in environmental samples. J Hazard Mater 168(2-3):1022–1027
Guha SN, Mittal JP (1992) Pulse radiolysis study of redox reactions of Safranine T in sodiumdodecylsulphate (SDS) micellar medium. Proc Indian Acad Sci Chem Sci 104:497–507
Guha SN, Mittal JP (1997) Pulse radiolysis study of one electron reduction of Safranine T. J Chem Soc Faraday Trans 93:3647–3652
Gürkan R, Altunay N (2013) Determination of trace cadmium in nonalcoholic beverages by coupling cloud point extraction with spectrophotometry. Pol J Food Nutr 63:253–260
Horwitz W (2000) Official method of analysis. 17th ed. Gaithersburg (MD, USA): Association of Analytical Chemists
Hosseini MS, Hamid HM (2005) Flotation-spectrophotometric determination of Ag(I) at the 10−7 mol L−1 level using iodide and Ferroin as an ion-associate. Bull Korean Chem Soc 26:1529–1532
Hu Q, Yang G, Zhao Y, Yin J (2003) Determination of copper, nickel, cobalt, silver, lead, cadmium, and mercury ions in water by solid-phase extraction and the RP-HPLC with UV-Vis detection. Anal Bioanal Chem 375(6):831–835
Kaličanin BM (2009) Determination of very toxic metal-cadmium in natural water samples. Desalination 249(1):58–62
Khammas ZAA, Ghali AA, Kadhim KH (2012) Combined cloud point extraction spectrophotometric extraction and spectrometric detection of lead and cadmium in honey samples using a new ligand. Int J Chem Sci 10(3):1185–1204
Krizkova S, Adam V, Kizek R (2009a) Phytotoxicity of silver ions. Chem Listy 103:559–568
Krizkova S, Krystofova O, Trnkova L, Hubalek J, Adam V, Beklova M, Kizek R (2009b) Silver (I) ions ultrasensitive detection at carbon electrodes-analysis of waters, tobacco cells and fish tissues. Sensors 9(9):6934–6950
López-García I, Vicente-Martínez Y, Hernández-Córdoba M (2014) Speciation of silver nanoparticles and Ag(I) species using cloud point extraction followed by electrothermal atomic absorption spectrometry. Spectrochim Acta B 101:93–97
López-García I, Vicente-Martínez Y, Hernández-Córdoba M (2015) Cloud point extraction assisted by silver nanoparticles for the determination of traces of cadmium using electrothermal atomic absorption spectrometry. J Anal At Spectrom 30:375–380
Mashhadizadeh M, Amoli-Diva M, Shapouri MR, Afruzi H (2014) Solid phase extraction of trace amounts of silver, cadmium, copper, mercury, and lead in various food samples based on ethylene glycol bis-mercaptoacetate modified 3-(trimethoxysilyl)-1-propanethiol coated Fe3O4 nanoparticles. Food Chem 151:300–305
Naeemullah KTG, Tuzen M (2016) Development of novel simultaneous single step and multistep cloud point extraction method for silver, cadmium and nickel in water samples. J Ind Eng Chem 35:93–98
Nardi EP, Evangelista FS, Tormen L, Saint TD, Curtius AJ, Souza SS, Barbosa F (2009) The use of inductively coupled plasma mass spectrometry (ICP-MS) for the determination of toxic and essential elements in different types of food samples. Food Chem 112(3):727–732
Olmedo P, Pla A, Hernández AF, Barbier F, Ayouni L, Gil F (2013) Determination of toxic elements (mercury, cadmium, lead, tin and arsenic) in fish and shellfish samples. Risk assessment for the consumers. Environ Int 59:63–72
Rahman MA, Kaneco S, Amin MN, Suzuki T, Ohta K (2004) Determination of silver in environmental samples by tungsten wire preconcentration method–electrothermal atomic absorption spectrometry. Talanta 62(5):1047–1050
Shirmardi-Dezaki A, Shamsipur M, Akhond M, Sharghi H, Doroodmand MM (2012) Array of potentiometric sensors for simultaneous determination of copper, silver, and cadmium ions in complex mixtures. Electrochim Acta 62:84–90
Silva EL, Roldan PDS, Giné MF (2009) Simultaneous preconcentration of copper, zinc, cadmium, and nickel in water samples by cloud point extraction using 4-(2-pyridylazo)-resorcinol and their determination by inductively coupled plasma optic emission spectrometry. J Hazard Mater 171:1133–1138
Silva DG, Junior MM, Silva LO, Portugal LA, Matos GD, Ferreira SL (2011) Determination of cadmium in rice by electrothermal atomic absorption spectrometry using aluminum as permanent modifier. Anal Methods 3(11):2495–2500
Silva FLF, Matos WO, Lopes GS (2015) Determination of cadmium, cobalt, copper, lead, nickel and zinc contents in saline produced water from the petroleum industry by ICP-OES after cloud point extraction. Anal Methods 7(23):9844–9849
Sosa-Ferrera Z, Padron-Sanz C, Mahugo-Santana C, Santana-Rodríguez JJ (2004) The use of micellar systems in the extraction and pre-concentration of organic pollutants in environmental samples. TRAC-Trend Anal Chem 23:469–479
Yang G, Fen W, Lei C, Xiao W, Sun H (2009) Study on solid phase extraction and graphite furnace atomic absorption spectrometry for the determination of nickel, silver, cobalt, copper, cadmium and lead with MCI GEL CHP 20Y as sorbent. J Hazard Mater 162(1):44–49
Zhu X, Zhu X, Wang B (2006) Determination of trace cadmium in water samples by graphite furnace atomic absorption spectrometry after cloud point extraction. Microchim Acta 154:95–100
Acknowledgments
Thanks are also due to Prof. Dr. Mehmet Akçay for his meaningful helps and contributions in critical evaluation and publication step of the presented research article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors have no financial relationship with the organization that sponsored the research.
Conflict of Interest
Ramazan Gürkan declares that he has no conflict of interest. Nail Altunay declares that he has no conflict of interest. Emre Yıldırım declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human or animal subjects.
Informed Consent
On behalf of other authors, informed consent was obtained from all individual participants included in the study.
Additional information
Highlights
✓ The detection limit of method is 0.02 and 0.9 μg L−1 for Ag(I) and Cd(II), respectively.
✓ The method is simple, low cost, and sensitive in accordance with the green chemistry concept.
✓ The parameters affecting UA-CPE efficiency were optimized before detection by FAAS.
✓ The method enables the determination of Ag(I) and Cd(II) in dried nuts and vegetables.
✓ The method shows satisfactory recovery and relative standard deviation values.
Rights and permissions
About this article

Cite this article
Gürkan, R., Altunay, N. & Yıldırım, E. Combination of Ultrasonic-Assisted Cloud Point Extraction with Flame AAS for Preconcentration and Determination of Trace Amounts of Silver and Cadmium in Dried Nut and Vegetable Samples. Food Anal. Methods 9, 3218–3229 (2016). https://doi.org/10.1007/s12161-016-0505-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-016-0505-7