
Abstract
In this study, three QuEChERS-based methods of improving sample extract preparation through reduction of unwanted matrix background for pesticide residue analysis in dill (a chlorophyll-containing matrix) were developed and compared. These methods involved the use of (1) dispersive solid-phase extraction (dispersive-SPE), (2) dispersive-SPE combined with liquid–liquid extraction (LLE), and (3) only LLE. Samples were analyzed by gas chromatography with electron capture and nitrogen phosphorus detection (GC-ECD/NPD). The results indicated that cleanup with primary secondary amine (PSA) and graphitized carbon black (GCB) sorbents followed by final solvent exchange from acetonitrile to petroleum ether was the best alternative of the tested methods, resulting in the cleanest extracts. The matrix effect of dill on the most favorable method herein was not significant, and the calibration performed well, with R 2 ≥ 0.99. The overall recoveries at three spiking levels of 0.01, 0.1, and 1.0 mg kg−1 fell in the range between 73 and 110 % (95 % on average) with relative standard deviation (RSD) values equal to or lower than 12 % (5 % on average). Uncertainty for the studied pesticides ranged from 7 to 14 % (with the overall average uncertainty of 11 %). Analyses of real samples revealed the presence of pesticides unapproved for use on dill, as well as exceedances of the maximum residue levels (MRLs) for some pesticides. The samples with residue violation results were also confirmed by gas chromatography–tandem mass spectrometry (GC-MS/MS). The proposed method of sample preparation and chromatographic analysis is sufficient and can be applied with all certainty to determine pesticide residues in dill and other plants of high pigment content, i.e., chlorophyll.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dill (Anethum graveolens L.) probably originates from the region of Southwest Asia or Southeast Europe (Bailer et al. 2001). In many countries, dill has long been cultivated as a spice plant, and due to its unique chemical composition, it also has therapeutic properties (both dill leaves and seeds). Dill consumption could lower the risk of cancer (Peerakam et al. 2014) and reduce the level of cholesterolemia (Yazdanparast and Bahramikia 2008). This plant owes its therapeutic benefits to many bioactive ingredients, including monoterpines (limonene, carvone), flavonoids, and carotenoids, e.g., beta-carotene which is mainly found in the essential oil of dill (Jana and Shekhawat 2010; Jirovetz et al. 2003; Callan et al. 2007). This could be considered as a source of a natural antimicrobial whereas its extract could be considered as an alternative source of natural antioxidant (Singh et al. 2005). Besides this, the plant is very rich in minerals, particularly calcium, iron, and potassium (Özcan 2004), vitamin C, and chlorophylls (Peerakam et al. 2014; Lisiewska et al. 2006; Daly et al. 2010).
Similarly, as in the case of many other crops, dill is susceptible to pests and disease attacks. One of the most common methods of protecting plants and plant products from the effects of harmful organisms is the use of active substances in plant protection products (Regulation 2005). As long as pesticides will be commonly used in agriculture, it will be necessary to monitor products intended for consumption that could contain residues of the applied pesticides. Because of the potential health risk to consumers resulting from acute and/or chronic dietary exposure, maximum residue limits (MRLs) have been established for many pesticides in the EU (Cajka et al. 2012). MRLs should be set at the lowest achievable level consistent with good agricultural practice for each pesticide with a view to protecting vulnerable groups such as infants and young children (Regulation EC 2005). A number of active substances are banned from being placed on the market and used in plant protection products (Regulation 2009). At the same time, the presence of certain active substances in plant products is the result of improper pesticide use for individual crops or environmental pollution. Considering all of the aforementioned aspects, plant products are analyzed for the presence of pesticide residues in order to ensure that they are suitable for consumption.
The choice of methodology for determining pesticides depends in large measure on the sample matrix and the structure and properties of the target analytes. In view of the numerous legal regulations laying down the highest permissible levels of pesticides in various matrices, sensitive and selective analytical techniques are used, appropriate to the low concentrations at which the target analytes occur in them. In addition, each stage in the analytical procedure, as well as this process in its entirety, should be validated (Beyer and Biziuk 2007; Namieśnik and Górecki 2001; Stocka et al. 2011). The complete procedure for determining pesticide residues in biological materials is complex and consists of several stages, which are preparation of samples for analysis (homogenization, separation into analytical portions, and adequate sample storage), extraction of pesticides from sample and/or enrichment of the sample, extract purification (the extract that is obtained requires removal of co-extracted ingredients like sugars, acids, and pigments, which could disrupt the results of analysis), identification, and determination of analytes (Fenik et al. 2011). Techniques such as solid-phase extraction (SPE), solid-phase micro-extraction (SPME) (Ruiz del Castillo et al. 2012), and, more recently, QuEChERS (Lehotay 2011), made possible to solve the drawbacks of other extraction techniques, making the experimental approach faster and environmentally friendly. The full range of extraction techniques now encompasses other types of methods: super-critical fluid extraction (SFE) (Ono et al. 2006), matrix solid-phase dispersion (MSDP) (Ramos et al. 2009), single-drop micro-extraction (SDME) (Pakade and Tewary 2010), stir bar sorptive extraction (SBSE) (Juan-Garćıa et al. 2004), pressurized liquid extraction (PLE) (Yarita et al. 2012), and microwave-assisted extraction (MAE) (Satpathy et al. 2011) which is, however, less prevalent. The typical techniques for determining pesticide residues in plant products are capillary gas chromatography (GC) and high-performance liquid chromatography (HPLC). The evolution of chromatographic analysis follows a path that led to the appearance of devices with a mass spectrometry (MS) detector, tandem mass spectrometry (MS-MS) as an operation mode, and time-of-flight mass spectrometry (TOF-MS) (Fernandes et al. 2011).
In this study, the multiresidue method based on the application of a modified QuEChERS method followed by gas chromatography coupled with electron capture and nitrogen phosphorus detection (GC-ECD/NPD) and confirmation of samples with residue violation results by gas chromatography–tandem mass spectrometry (GC-MS/MS) were used for the analysis of 22 pesticides in dill. Method validation in terms of recovery, precision, and linearity, as well as estimate of matrix effects and measurement uncertainty, is reported. It was verified whether methods of improving sample preparation achieved their intended objective—acceptable recovery and repeatability of the target analytes and reduction of the amount of co-extractives, simultaneously. In order to demonstrate fitness for purpose of the developed and validated method, it was applied to the determination of pesticide residues in real dill samples.
Material and Methods
Chemical and Reagents
All solvents for pesticide residue analysis were of high purity. Acetonitrile and petroleum ether were obtained from Honeywell Specialty Chemicals Seelze GmbH (Germany), and acetone was obtained from Honeywell (USA). Dispersive SPE sorbents for method experiments (pre-weighed mixtures of 4 g anhydrous magnesium sulphate, 1 g sodium chloride, 0.5 g di-sodium hydrogen citrate sesquihydrate, and 1 g sodium citrate dehydrate; pre-weighed mixtures of 150 mg primary secondary amine (PSA), 45 mg graphitized carbon black (GCB), and 900 mg anhydrous magnesium sulfate) were purchased from Perlan Technologies (Poland); sodium chloride (NaCl) was from Chempur (Poland).
Pesticide Analytical Standards
Pesticide standards, all 95 % or of higher purity, were purchased from LGC Standards, formerly Dr. Ehrenstorfer (Germany). Individual pesticide stock solutions were prepared at approximate concentrations of 1000 μg mL−1 in analytical grade acetone and stored in closed bottles at ≤−16 °C. A single composite pesticide mixture at approximately 10 μg mL−1 was prepared from the individual stock solutions by dilution with analytical grade acetone. Subsequent working standards were prepared by dilutions of the appropriate volumes of the pesticide mixture with acetone. Matrix-matched standards were acquired by mixing working standard solutions with dill blank sample extracts (in petroleum ether) containing 0.5 g sample per 1 mL solvent. All working standard solutions were stored at 4 °C in dark amber bottle for further analysis.
Samples
Samples of fresh dill for the use in spiking experiments were purchased from a local organic farm. Before the recovery tests, the samples were verified for the absence of pesticide residues and of interferences instrumentation and conditions for chromatographic analysis. Dill samples were used for blanks, spiking samples for recovery assays, and preparation of matrix-matched standards for calibration purposes. Samples for the monitoring study (n = 18) were purchased from the local markets.
Sample Preparation and Cleanup Procedure
Sample preparation was based on the QuEChERS method (Anastassiades et al. 2003) and has been modified as follows: the sample size was reduced to 5 g, 10 mL water was added, and solvent was exchanged from acetonitrile to petroleum ether before GC-ECD and GC-NPD analysis. The water was added before proceeding with the acetonitrile extraction to enable the solvent to penetrate better the plant tissues and ensure complete transfer of the analytes.
Extraction
Five grams of homogenized sample was weighed into a polypropylene centrifuge tube (50 mL), and 10 mL acetonitrile and 10 mL distilled water were added. The contents were shaken manually for 2 min, and then a mixture of 1 g sodium chloride, 0.5 g disodium hydrogen citrate sesquihydrate, 1 g sodium citrate dehydrate, and 4 g magnesium sulfate was added. Again, the contents were shaken manually for 1 min and centrifuged at 3000 rpm for 5 min.
Cleanup
Sample extract purification was performed according to three different methods:
-
(A)
Dispersive SPE
A 6-mL aliquot of the upper layer was transferred into a 15-mL disposable polypropylene centrifuge tube with pre-weighed 150 mg PSA, 45 mg GCB, and 900 mg anhydrous magnesium sulfate. The contents were shaken manually for 2 min and centrifuged at 3000 rpm for 5 min.
For analysis by GC-ECD/NPD, a 2-mL aliquot of the supernatant was evaporated under a gentle stream of nitrogen and reconstituted in 2 mL of petroleum ether.
-
(B)
Dispersive SPE + LLE
The procedure was the same as in (A), but the next step was to perform liquid–liquid extraction (LLE) by using 2 mL acetonitrile extract, 2 mL petroleum ether, and 10 mL 20 % aqueous NaCl solutions. The contents were shaken manually for 1 min, and the upper ether extract was used for gas chromatography analysis.
-
(C)
LLE only
Two milliliters of the crude extract was taken and subjected to the LLE extraction procedure as in (B). The upper ether extract was used for gas chromatographic analysis.
Instrumentation and Conditions for Chromatographic Analysis
An Agilent Technologies 6890 GC gas chromatograph (USA), HP ChemStation, Rev. A 10.02 equipped with electronic pressure control (EPC) and an autosampler was used to detect the residue of pesticide in dill. Analytes were separated on a DB-1701 30 m × 0.25 mm × 0.25 μm capillary column (Agilent Technologies, USA). Nitrogen of 6.0 purity was used as the carrier gas. The column was connected to the nitrogen phosphorus detector (NPD) and the electron capture detector (ECD) using a universal Y-splitter. The ECD and NPD were maintained at 270 and 300 °C, respectively. Nitrogen was the makeup gas for the ECD (30 mL min−1). For the NPD, hydrogen and air flows were kept at 3 and 30 mL min−1, respectively. The makeup gas was nitrogen at 30 mL min−1. The column temperature program was as follows: 100 °C (1 min) → 20 °C/min → 180 °C (4 min) → 20 °C/min → 220 °C (5 min) → 20 °C/min → 260 °C (48 min). The injector temperature was held at 250 °C. Sample extract volumes of 2 μL were injected in splitless mode. Total run time was 65 min. In addition to that, the samples containing pesticides above MRL levels or not approved for use on dill could be confirmed by GC-MS/MS. The instrumental conditions used for the GC-MS/MS confirmatory analyses are detailed in Supplementary information provided with this article.
Validation of the Method
The objective of validating the analytical method was to determine whether the process of analysis according to the given method is dependable and yields reliable results. The developed method was subjected to validation study using green parts of the dill (samples were previously checked to be free of the target pesticides and interferences).
Selectivity was tested by analyzing GC-ECD/NPD chromatograms of a blank and a fortified sample, verifying for interferences at the same retention time of the pesticides. Accuracy and precision (calculated from the recovery experiments, expressed as relative standard deviation, RSD) were determined by analyzing replicate (n = 5) samples fortified at three levels: 0.01, 0.1, and 1 mg kg−1. The samples were spiked before the sample preparation procedure was performed.
The method’s limit of quantification (LOQ) was determined as the lowest concentration that could be quantified with acceptable recovery (70–120 %) and precision (RSD ≤20 %) (Document SANCO 2013).
Linearity of calibration curves was studied by GC-ECD/NPD analysis at five concentration levels, 0.01, 0.05, 0.1, 0.5, and 1.0 μg mL−1, and they were applied in three repetitions per level, and response versus concentration curves were plotted. Repeatability precision was expressed as the relative standard deviation (RSD %). Calibration standards were prepared in petroleum ether (in pure solvent) and in dill extracts for comparison purposes and matrix effect magnitude evaluation. Matrix effect (ME) was calculated for each of 22 analytes by using the formula: % ME = [(slope of matrix-matched calibration curve − slope of reagent-only calibration curve) / slope of reagent-only calibration curve] × 100 (Kwona et al. 2012; Słowik-Borowiec 2015).
The measurement uncertainty was estimated based on the data obtained in the validation study. The major uncertainty sources included in the uncertainty budget were the repeatability of recoveries from spiked samples and uncertainty of the average recovery calculated from rectangular distribution. The relative expanded uncertainty was calculated by using the coverage factor k = 2 at the confidence level of 95 % (Medina-Pastor et al. 2011; Walorczyk and Drożdżyński 2012).
Results and Discussion
In the first step, 22 pesticides were selected, representing various chemical groups and characterized by a wide variety of physico-chemical properties (11 were insecticides, 9 fungicides, and 2 herbicides) that could potentially have been or continue to be used for protection of dill crops against pests and diseases. Also, the pesticides detected in recent years in the group of herbs and spices in the Rapid Alert System for Food and Feed (RASFF 2014) were included in the scope this work (namely, bifenthrin, chlorpyrifos, cypermethrin, flusilazole, phosalone, and propiconazole.
Sample Preparation
Above all, due to the large amount of pigments, i.e., chlorophyll and other ingredients mentioned in the first section, dill has a complex matrix, which is why the selection of sorbents such as GCB and PSA for the sample extract preparation step was acknowledged as necessary (Lehotay et al. 2010). It was also investigated whether the application and additional liquid--liquid extraction (LLE) step at the end of the analytical process would give better results (Cajka et al. 2012). The effectiveness of purification by sorbents and LLE was verified based on the presence of interfering peaks on the chromatogram as well as on recovery values, and sample discoloration was also accounted for.
Three different methods of purifying dill samples—(1) dispersive-SPE followed by final solvent exchange from acetonitrile to petroleum ether (A), (2) combination of dispersive-SPE and LLE (B), and (3) purification with only LLE (C)—were evaluated, obtained extracts were injected into the gas chromatograph, and chromatograms were compared for better cleanup in the form of reduced background and lower level of co-extracted interferences visible on the chromatograms.
For recovery and RSD study, standards were in matrix extracts prepared in the same way like samples.
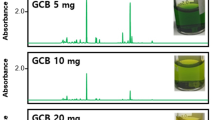
The most effective removal of matrix co-extractives, which translates to improvement of sensitivity, can be observed in the case method (A), i.e., dispersive-SPE followed by evaporation under a N2 stream and solvent exchange with petroleum ether and (B), i.e., dispersive-SPE and additional extraction with petroleum ether and 20 % NaCl. The extract color distinctly changed to bright yellow in these cases. The poorest purification was achieved for method (C) which involved only extraction with petroleum ether and 20 % NaCl. The extract remained green in color. Figures 1 and 2 present chromatograms from EC and NP detectors for the three extract purification techniques.

GC-ECD chromatograms of extracts of dill samples prepared and cleaned up by a dispersive-SPE with sorbents PSA and GCB; b dispersive-SPE with sorbents PSA and GCB, and then liquid–liquid extraction (LLE) using petroleum ether and 20 % NaCl; and c liquid–liquid extraction (LLE) using petroleum ether and 20 % NaCl

GC-NPD chromatograms of extracts of dill samples prepared and cleaned up by a dispersive-SPE with sorbents PSA and GCB; b dispersive-SPE with sorbents PSA and GCB, and then liquid–liquid extraction (LLE) using petroleum ether and 20 % NaCl; and c liquid–liquid extraction (LLE) using petroleum ether and 20 % NaCl
Thereafter, the recoveries from fortified samples by using the three evaluated methods were determined and compared. On the other hand, the method yielding the most effective removal of matrix co-extractives, i.e., method (A), was subjected to full validation study in terms of evaluation of linearity and matrix effects, recovery, and precision, as well as estimation of measurement uncertainty.
Linearity and Matrix Effects
Linearity was evaluated by studying five-level calibration curves plotted for pesticide standards prepared in solvent (petroleum ether) as well as in dill extracts obtained by dispersive-SPE followed by final solvent exchange from acetonitrile to petroleum ether, over a concentration range of 0.01–1.0 mg kg−1. The linearity parameters were highly satisfactory with coefficients of determination (R 2) ≥0.99 for all the tested analytes. Detailed linearity data (equations and R 2 values) can be found in Supplementary information included with this article.
For the purpose of estimating matrix effects, differences between the slope of the matrix-matched and solvent-only calibration curves were calculated and divided by the slope of the solvent-only calibration curve (Kwona et al. 2012). Calculated % MEs of the studied compounds are presented graphically in Fig. 3. The relative standard deviation (RSD) for the injections of these calibration curves in three repetitions was within the range from 0.02 to 11.8 %. Percent ME values were in the range of −5.2 % (bifenthrin) and 7.5 % (azoxystrobin) and were very small for the vast majority of the pesticides, 2.6 % on average, showing weak enhancement. To obtain accurate results for the pesticide residue analysis, quantifications should be done using matrix-matched calibration.

Matrix effects of target pesticides for GC-ECD/NPD analysis of dill extracts cleaned up by dispersive-SPE with sorbents PSA and GCB (method (A))
Walorczyk et al. analyzed 25 pesticides in chlorophyll-containing samples including green plants of lupin, white mustard, and sorghum by using QuEChERS with PSA/ChloroFiltr cleanup followed by GC-MS/MS or UPLC-MS/MS determination. The matrix effects occurred in large extent from −116 to 82 %. Strong matrix effects with absolute values >20 % were exhibited by the majority of pesticide and matrix combinations (>80 %) (Walorczyk et al. 2015). Therefore, the results obtained in the present work can be considered as an improvement over existing methods in terms of reduction of matrix effects in analysis of chlorophyll-containing matrices.
Recovery Study and Measurement Uncertainty
Trueness and precision in terms of average recovery and RSD were evaluated for the proposed methods by conducting recovery experiments at two dill sample enrichment levels (0.01 and 1.0 mg kg−1) and at three enrichment levels (0.01, 0.1, and 1.0 mg kg−1) in the case of the selected most favorable method (A) entailing dispersive SPE followed by final solvent exchange from acetonitrile to petroleum ether. The recovery and repeatability RSD values were determined by the analysis of five replicate spiked samples at each level, using matrix-matched standards prepared in dill extracts. According to the criteria contained in the SANCO/12571/2013 EU method validation procedure, average recovery should be between 70 and 120 % and RSD less than or equal to 20 %. The lowest spiking level corresponded to the limit of quantification (LOQ), and it was 0.01 mg kg−1. The determined LOQ was less than or equal to established MRLs (Regulation 2005) of the substances tested for in dill.
Conducted studies showed that the best recoveries (73–110 %, 95 % on average) and %RSD (0–12 %, 5 % on average) were obtained in method (A) (dispersive SPE followed by final solvent exchange from acetonitrile to petroleum ether) at three enrichment levels of 0.01, 0.1, and 1.0 mg kg−1. Full validation data achieved by using this method are detailed in Table 1.
In the second purification variant (B), combination of dispersive-SPE and LLE, recovery values were more varied (Fig. 4.): 88–124 %, RSD 1–14 % (level, 1.0 mg kg−1), and 82–127 %, RDS 1–21 % (level, 0.01 mg kg−1).

Average recoveries and RSDs for GC-ECD/NPD determination of target pesticide residues in dill; cleanup by dispersive-SPE with sorbents PSA and GCB, and then liquid–liquid extraction (LLE) using petroleum ether and 20 % NaCl at the two fortification levels (method (B))
In the third method (C), involving purification with only LLE, recovery values were within the range of 66–122 % with RSD equal to 6–20 % (level, 1.0 mg kg−1) and 71–193 %, RSD 1–62 % (level, 0.01 mg kg−1) (Fig. 5). Of the pesticides under study, tecnazene exhibited the highest recovery rate of 193 % and the poorest repeatability RSD of 62 % probably due to the influence of co-extracted matrix components.

Average recoveries and RSDs for GC-ECD/NPD determination of target pesticides residues in dill; cleanup by liquid–liquid extraction (LLE) using petroleum ether and 20 % NaCl at the two fortification levels ((method (C)))
A comparison of obtained results showed that the third purification variant has the greatest impact on the recovery rate, particularly at a lower enrichment level, because most tested analytes did not fulfill required SANCO (2013) criteria. The second purification method that was presented, involving a combination of dispersive-SPE and LLE, achieved satisfactory results that exceed acceptable limits only slightly. The results of experiments prove that the use of PSA and GCB sorbents is indispensable to the removal of co-extracts (also clearly visible as discoloration of the solution), and the use of just LLE with the omission of dispersive-SPE is not sufficient for this process. A 20 % aqueous NaCl solution was used in LLE to enhance the transfer of compounds into the petroleum layer by means of the salting out effect (Cajka et al. 2012). According to this author, the application of this technique to raw acetonitrile extracts is beneficial and gives the same results as in the case of two-step purification (dispersive-SPE and LLE) but with less sample manipulation steps.
There are several approaches to estimating measurement uncertainty at pesticide residue testing laboratories, primarily (1) based on experimental data from the quality control work of the laboratory pertaining to laboratory reproducibility, interlaboratory validation, or a combination of results obtained in proficiency tests or (2) identification of all possible sources of uncertainty in the entire analytical process and calculation of the uncertainty linked to each of them, which proves to be particularly time-consuming (Medina-Pastor et al. 2011; Eurachem/CITAC guide 2012). By taking the second approach to uncertainty estimation in our study, we obtained results indicating that repeatability and uncertainty of extraction recovery are the most important sources of uncertainty, and other sources, such as uncertainties of weighing and diluting standards, uncertainties of purity of standards, or matrix effect, only account for less than 1 % in our method. Expanded uncertainty was calculated by multiplying the combined standard uncertainty by expansion coefficient k. Probability was accepted at 95 %, with an expansion coefficient of k = 2. The results pertaining to technique (B) have a somewhat wider range, between 8 % (boscalid) and 26 % (trifloxystrobin), 17 % on average. The highest uncertainty values were obtained for technique (C) from 22 % (pyrimethanil) to 49 % (trifloxystrobin, tecnazen, and pirimicarb). The highest uncertainty value was obtained for compounds with high or poor recovery and high RSD value.
The results pertaining to method (A) of extract purification (i.e., dispersive-SPE followed by final solvent exchange from acetonitrile to petroleum ether), which was found to be the most favorable of the evaluated methods in terms of analytical performance parameters, are presented in Table 1. As shown by the data, uncertainty for the target pesticides ranged from 7 to 14 % with an overall average uncertainty of 11 %. This was distinctively less than a default value of ±50 % recommended by the EU SANCO/12571/2013 guidance document, demonstrating suitability of the method for the intended application.
Application to Real Samples
To demonstrate ruggedness of the selected method of the three evaluated ones, it was used to analyze 18 samples of fresh dill originating from local shops. The test results were interpreted in accordance with the criteria accepted in the European Union SANCO/12571/2013 guidance document (Document SANCO 2013), as well as by comparison with the MRLs currently enforced in Poland (Regulation 2005). Verification of correct application of pesticides was conducted on the basis of the current “Register of plant protection products approved for marketing and application” and “Label-instructions for use on plant protection products approved for marketing and application with a permit of the Ministry of Agriculture and Rural Development” (Register 2015; Label-instructions 2015). The test results are presented in Table 2, where the MRLs of substances detected in analyzed dill samples are also given. Most of the samples (83 %) did not contain any pesticide residues. In three samples, pesticide residues were found separately. But, chlorpyrifos was detected in one sample in a quantity exceeding the MRLs, and active substances of pesticide preparations banned from use for dill protection were detected (these preparations could still have legally been used in past years; permit was revoked). In other one sample, pendimethalin which is not officially approved for use on dill in Poland was detected. The samples with residue violation results were also confirmed by gas chromatography–tandem mass spectrometry (GC-MS/MS). Figure 6 shows the confirmation of pendimethalin identity in a dill sample by measurement of two transitions of precursor ion fragmenting to product ions at the proper ion ratio, as recommended by the SANCO/12571/2013 guidance document.

GC-MS/MS MRM chromatograms showing confirmation of the identity of pendimethalin residues is a dill sample
Dill belongs to the group of “minor crops,” and the main problem in crops of this type is the very low number of agents registered for their protection. These plants, similarly to many others, are susceptible to attacks by pests and pathogens, so they require the application of pesticides. The lack of registered preparations is the reason why unapproved preparations are being used, and this leads to cases where the law is violated.
Conclusions
The three modified QuEChERS-based multiresidue methods for the analysis of 22 pesticides in dill using GC–ECD/NPD were evaluated and compared. The results of conducted studies indicated that purification with the application of mixed-sorbent cleanup (PSA and GCB), followed by solvent exchange by evaporation in an N2 stream, yields the best results. The purest sample extracts were obtained without major matrix effects (−5.2 to 7.5 %), which significantly improved quantification. Satisfactory results of modified QuEChERS verification were also obtained in terms of precision, recovery, and uncertainty. The presented method of sample preparation and chromatographic analysis is sufficient, and it can be applied with all certainty to determine a wide range of pesticides in dill and other plants of high pigment content, i.e., chlorophyll. The second and third purification variant for some tested analytes did not fulfill the required SANCO (2013) criteria.
Analyses of real samples revealed the presence of pesticides unapproved for the use on dill, as well as a case of violation of the pesticide MRL. The obtained results indicate the need to monitor pesticide residues in dill crops or other “minor crops” or herb crops.
References
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86:412–431
Bailer J, Aichinger T, Hackl G, Hueber KD, Dachler M (2001) Essential oil content and composition in commercially available dill cultivars in comparison to caraway. Indus Crop Prods 14:229–239
Beyer A, Biziuk M (2007) Przegląd metod oznaczania pozostałości pestycydów i polichlorowanych bifenyli w próbkach żywności. Ecol Chem Eng 14:35–58 [in Polish]
Cajka T, Sandy C, Bachanova V, Drabova L, Kalachova K, Pulkrabova J, Hajslova J (2012) Streamlining sample preparation and gas chromatography–tandem mass spectrometry analysis of multiple pesticide residues in tea. Anal Chim Acta 743:51–60. doi:10.1016/j.aca.2012.06.051
Callan NW, Johnson DL, Westcott MP, Welty LE (2007) Herb and oil composition of dill (Anethum graveolens L.): effects of crop maturity and plant density. Ind Crop Prod 25:282–287. doi:10.1016/j.indcrop.2006.12.007
Daly T, Jiwan MA, O’Brien NM, Aherne SA (2010) Carotenoid content of commonly consumed herbs and assessment of their bioaccessibility using an in vitro digestion model. Plant Food Hum Nutr 65:164–169. doi:10.1007/s11130-010-0167-3
del Castillo ML R, Rodriguez-Valenciano M, De la Peña Moreno F, Blanch GP (2012) Evaluation of pesticide residue contents in fruit juice by solid-phase microextraction and multidimensional gas chromatography coupled with mass spectrometry. Talanta 89:77–83, S0039-9140(11)01044-7
Document SANCO/12571/2013. Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. pp. 42.
Label-instructions (2015) Label-instructions for use on plant protection products approved for marketing and application with a permit of the Ministry of Agriculture and Rural Development.https://bip.minrol.gov.pl/Informacje-Branzowe/Produkcja-Roslinna/Ochrona-Roslin/Etykiety-Srodkow-Ochrony-Roslin?%2FInformacje-Branzowe%2FProdukcja-Roslinna%2FOchrona-Roslin%2FEtykiety-Instrukcji-Stosowania-Srodkow-Ochrony-Roslin. Accessed 14 April 2015
Eurachem/CITAC guide: Quantifying Uncertainty in Analytical Measurement (2012) Ellison SLR, Williams A. (Eds) Third edition ISBN 978-0-948926-30-3. Available from www.eurachem.org.
Fenik J, Tankiewicz M, Biziuk M (2011) Properties and determination of pesticides in fruits and vegetables. Trends Anal Chem; TrAC 30:814–826. doi:10.1016/j.trac.2011.02.008
Fernandes VC, Domingues VF, Mateus N, Delerue-Mato C (2011) Determination of pesticides in fruit and fruit juices by chromatographic methods. An overview. J Chromatogr Sci 49:715–730
Jana S, Shekhawat GS (2010) Anethum graveolens, an Indian traditional medicinal herb and spice. Pharmacogn Rev 4:179–184. doi:10.4103/0973-7847.70915
Jirovetz L, Buchbauer G, Stoyanova AS, Georgiev EV, Damianova ST (2003) Composition, quality control, and antimicrobial activity of the essential oil of long-time stored dill (Anethum graveolens L.) seeds from Bulgaria. J Agric Food Chem 51:3854–3857. doi:10.1021/jf030004y
Juan-Garćıa A, Mãnes J, Font G, Pićo Y (2004) Evaluation of solid-phase extraction and stir-bar sorptive extraction for the determination of fungicide residues at low-microg kg(−1) levels in grapes by liquid chromatography-mass spectrometry. J Chromatogr A 1050:119–127
Kwona H, Lehotay SJ, Geis-Asteggiante L (2012) Variability of matrix effects in liquid and gas chromatography–mass spectrometry analysis of pesticide residues after QuEChERS sample preparation of different food crops. J Chromatogr A 1270:235–245. doi:10.1016/j.chroma.2012.10.059
Lehotay SJ (2011) QuEChERS sample preparation approach for mass spectrometric analysis of pesticide residues in foods. Methods Mol Biol 747:65–91. doi:10.1007/978-1-61779-136-9_4
Lehotay SJ, Son KA, Kwon H, Koesukwiwat U, Fu W, Mastovska K, Hoh E, Leepipatpiboon N (2010) Comparison of QuEChERS sample preparation methods for the analysis of pesticide residues in fruits and vegetables. J Chromatogr A 1217:2548–2560. doi:10.1016/j.chroma.2010.01.044
Lisiewska Z, Kmiecik W, Korus A (2006) Content of vitamin C, carotenoids, chlorophylls and polyphenols ingreen parts of dill (Anethum graveolensL.) depending on plant height. J Food Comp Anal 19:134–140. doi:10.1016/j.jfca.2005.04.009
Medina-Pastor P, Valverde A, Pihlström T, Masselter S, Gamon M, Mezcua M, Rodríguez-Torreblanca C, Fernández-Alba AR (2011) Comparative study of the main top-down approaches for the estimation of measurement uncertainty in multiresidue analysis of pesticides in fruits and vegetables. J Agric Food Chem 59:7609–7619. doi:10.1021/jf104060h
Namieśnik J, Górecki T (2001) Preparation of environmental samples for the determination of trace constituents. Pol J Environ Stud 10:77–84
Ono Y, Yamagami T, Nishina T, Tobino T (2006) Pesticide multiresidue analysis of 303 compounds usings fluid extraction. Anal Sci 22:1473–1776. doi:10.2116/analsci.22.1473
Özcan M (2004) Mineral contents of some plants used as condiments in Turkey. Food Chem 84:437–440. doi:10.1016/S0308-8146(03)00263-2
Pakade YB, Tewary DK (2010) Development and applications of single-drop microextraction for pesticide residue analysis: a review. J Sep Sci 33:3683–3691. doi:10.1002/jssc.201000331
Peerakam N, Wattanathorn J, Punjaisee S, Buamongkol S, Sirisa-ard P, Chansakaow S (2014) Chemical profiling of essential oil composition and biological evaluation of Anethum graveolens L. (seed) grown in Thailand. Journal of Natural Sciences Research ISSN 2224–3186 (Paper) ISSN 2225–0921 (Online) 4 (16).
Ramos JJ, González MJ, Ramos L (2009) Comparison of gas chromatography-based approaches after fast miniaturized sample preparation for the monitoring of selected pesticide classes in fruits. J Chromatogr A 1216:7307–7313. doi:10.1016/j.chroma.2009.05.013
Rapid Alert System for Food and Feed (RASFF) (2014) https://webgate.ec.europa.eu/rasff-window/portal/?event=SearchForm&cleanSearch=1#. Accessed 14 Oct 2014
Regulation (EC) No 1107/2009 of the European Parliament and of the Council of 21 October 2009 concerning the placing of plant protection products on the market and repealing Council Directives 79/117/EEC and 91/414/EEC Official Journal of the European Union L 309/1 24.11.2009. pp. 50.
Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin and amending Council Directive 91/414/EEC. Off. J. L 70, with later amendments. pp. 16 http://ec.europa.eu/sanco_pesticides/public/?event=product.resultat&language=PL&selectedID=169. Accessed 17 Apr 2015
Register (2015) Register of plant protection products approved for marketing and application. https://bip.minrol.gov.pl/Informacje-Branzowe/Produkcja-Roslinna/Ochrona-Roslin/Rejestr-Srodkow-Ochrony-Roslin. Accessed 14 Apr 2015
Satpathy G, Tyagi YK, Gupta RK (2011) A novel optimised and validated method for analysis of multi-residues of pesticides in fruits and vegetables by microwave-assisted extraction (MAE)–dispersive solid-phase extraction (d-SPE)–retention time locked (RTL)–gas chromatography–mass spectrometry with deconvolution reporting software (DRS). Food Chem 127:1300–1308. doi:10.1016/j.foodchem.2011.01.087
Singh G, Maurya S, de Lampasona MP, Catalan C (2005) Chemical constituents, antimicrobial investigations, and antioxidative potentials of Anethum graveolens L. essential oil and acetone extract: Part 52. J Food Sci 70:M208–M215. doi:10.1111/j.1365-2621.2005.tb07190.x
Słowik-Borowiec M (2015) Validation of a QuEChERS-based gas chromatographic method for multiresidue pesticide analysis in fresh peppermint including studies of matrix effects. Food Anal Methods 8:1413–1424. doi:10.1007/s12161-014-0027-0
Stocka J, Tankiewicz M, Biziuk M, Namieśnik J (2011) Green aspects of techniques for the determination of currently used pesticides in environmental samples. Int J Mol Sci 12:7785–7805. doi:10.3390/ijms12117785
Walorczyk S, Drożdżyński D (2012) Improvement and extension to new analytes of a multi-residue method for the determination of pesticides in cereals and dry animal feed using gas chromatography–tandem quadrupole mass spectrometry revisited. J Chromatogr A 1251:219–231. doi:10.1016/j.chroma.2012.06.055
Walorczyk S, Drożdżyński D, Kierzek R (2015) Determination of pesticide residues in samples of minor drops by gas chromatography and ultra performance liquid chromatography coupled to tandem mass spectrometry. Talant 132:197–204
Yarita T, Aoyagi Y, Otake T (2012) Evaluation of pressurized liquid extraction for determination of organophosphorus and pyrethroid pesticides in soybean. J Environ Sci Health B 47:942–948. doi:10.1080/03601234.2012.706559
Yazdanparast R, Bahramikia S (2008) Evaluation of the effect of Anethum graveolens L. crude extracts on serum lipids and lipoproteins profiles in hypercholesterolaemic rats. DARU, J Pharm Sci 16:88–94
Acknowledgments
Magdalena Sƚowik-Borowiec is grateful to the Institute of Plant Protection – National Research Institute for her pre-doctoral scholarship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported in part by Ministerstwo Nauki i Szkolnictwa Wyższego (Ministry of Science and Higher Education), project ID: SRZ-08.
Conflict of Interest
Magdalena Słowik-Borowiec declares that she has no conflict of interest. Stanisław Walorczyk declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Rights and permissions
About this article

Cite this article
Słowik-Borowiec, M., Walorczyk, S. QuEChERS-Based Methods for the Determination of Pesticide Residues in a Dill Using Gas Chromatography with Electron Capture and Nitrogen Phosphorus Detection. Food Anal. Methods 9, 1562–1572 (2016). https://doi.org/10.1007/s12161-015-0333-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-015-0333-1